








Agammaglobulinämie
Agammaglobulinämien bei Hunden
ist eine Mangelerkrankung, bei der der Körper bestimmte Antikörper nicht ausbildet. Bisher wurde ein isolierter Fall von CID bekannt, der in einer Familie von Bassethunden auftrat. Eine schwerere Form der CID tritt auch bei Jack Russell Terriern auf.
ist eine Mangelerkrankung, bei der der Körper bestimmte Antikörper nicht ausbildet. Bisher wurde ein isolierter Fall von CID bekannt, der in einer Familie von Bassethunden auftrat. Eine schwerere Form der CID tritt auch bei Jack Russell Terriern auf.
eingestellt von: ise; Quellennachweis
Ataxie und Myelopathie der Terrier
Die Ataxie und Myelopathie der Terrier
(Hereditäre Ataxie) kommt bei Foxterrier und Jack Russell Terrier sowie Parson Russell Terrier vor. Hierbei tritt ein Abbau der weißen Substanz im Hals- und Brustbereich des Rückenmarks auf. Sie entwickelt sich im Alter von 2 bis 6 Monaten und geht mit weit ausladenden Bewegungen (Hypermetrie) der Vorderbeine, Muskelzittern und breitbeiniger Stellung der Hinterbeine einher.
Die Tiere neigen zum Umfallen und können nicht mehr alleine aufstehen. Beim Jack Russell bzw. Parson Russell Terrier kommt es auch zur Schädigung des Hörnervs mit zunehmender Taubheit.
(Hereditäre Ataxie) kommt bei Foxterrier und Jack Russell Terrier sowie Parson Russell Terrier vor. Hierbei tritt ein Abbau der weißen Substanz im Hals- und Brustbereich des Rückenmarks auf. Sie entwickelt sich im Alter von 2 bis 6 Monaten und geht mit weit ausladenden Bewegungen (Hypermetrie) der Vorderbeine, Muskelzittern und breitbeiniger Stellung der Hinterbeine einher.
Die Tiere neigen zum Umfallen und können nicht mehr alleine aufstehen. Beim Jack Russell bzw. Parson Russell Terrier kommt es auch zur Schädigung des Hörnervs mit zunehmender Taubheit.
eingestellt von: ise; Quellennachweis
Axonopathie des Labradors
Die Axonopathie des Labradors
ist eine bei Labrador Retrievern auftretende Degeneration der weißen Substanz. Sie beginnt mit Hinterhandschwäche bereits im Welpenalter und entwickelt sich zu ausgeprägten Hypermetrien mit Neigung zum Umfallen.
ist eine bei Labrador Retrievern auftretende Degeneration der weißen Substanz. Sie beginnt mit Hinterhandschwäche bereits im Welpenalter und entwickelt sich zu ausgeprägten Hypermetrien mit Neigung zum Umfallen.
eingestellt von: ise; Quellennachweis
Brachycephalie

Brachycephalie (auch: Brachyzephalie, von kurz und Kopf) bedeutet Kurzköpfigkeit bzw. Rundköpfigkeit. Es handelt sich dabei um eine angeborene, erbliche Deformation des Schädels, die zu verschiedenen gesundheitlichen Problemen führt. Unter den Haustieren sind insbesondere Hunde betroffen.
Die Definition eines Schädels als brachycephal geschieht nach mehreren Kriterien:
- Kurzer, breiter Gesichtsschädel
- Verhältnis Schädelbreite zu Schädellänge von 0,81 oder höher
- Verhältnis Hirnschädel zu Gesichtsschädel größer 1,6
- Kraniofazialwinkel zwischen 9° und 14°
Der Kraniofazialwinkel ist der von der Schnauzenspitze, der Schädelbasis und dem Mittelpunkt der Augenhöhle begrenzte Winkel.
Gesundheitliche Folgen
Brachycephales Syndrom
Brachycephalie kann zu Problemen der oberen Atemwege führen, die zusammenfassend als brachycephales Syndrom oder Brachycephalie-Syndrom bezeichnet werden. Es ist durch eine starke Behinderung der Atmung und eine gestörte Thermoregulation gekennzeichnet.
Pathogenese
Auslöser der Verengung der oberen Luftwege ist vor allem eine Hemmung des Längenwachstums des Gesichtsschädels. Durch das voluminöse Ausmaß des Knorpels des Nasenflügels (insbesondere die Cartilago nasi lateralis dorsalis) und einer reduzierten Beweglichkeit des Nasenflügels kommt es zu einer Verengung der Nasenlöcher.
Die Nasenhöhle des heranwachsenden Hundes streckt sich normalerweise in die Länge und die zur Geburt noch unterentwickelten Nasenmuscheln wachsen aufeinander zu, stellen aber ihr Wachstum ein bevor die sie überziehende Schleimhautschichten einander berühren. Bei brachycephalen Hunden unterbleibt diese Wachstumshemmung – es kommt zu einer relativen Conchen-Hypertrophie („Nasenmuschelwucherung“). Zudem wachsen die Nasenmuscheln als aberrante Nasenmuscheln über ein für die Größe der Nasenhöhle mögliches Maß.
Man unterscheidet dabei rostrale aberrante Nasenmuscheln (vor der Plica alaris gelegen), die vorderen Abschnitte des mittleren und unteren Nasengangs einengen, und kaudale aberrante Nasenmuscheln, die den Übergang zum Nasenrachen (Meatus nasopharyngeus) einengen. Zudem sind die Lamellen der Nasenmuscheln bei brachycephalen Hunden deutlich dicker als bei anderen Hunden. Die Folge ist, dass kaum noch Luft durch die Nase strömen kann, was für einen obligaten Nasenatmer ein schwerwiegendes Problem darstellt.
Das Gaumensegel brachycephaler Hunde ist verlängert und verdickt. Dies führt zu einer Einengung des Nasenrachens sowie in Kombination mit der relativ zu großen Zunge (relative Makroglossie) auch des Mundrachens.
Im Bereich des Kehlkopfs treten ebenfalls krankhafte Veränderungen auf, vor allem beim Mops. Hier ist das Kehlkopfskelett instabil, insbesondere der Stellknorpel und der Kehldeckel, so dass die Gefahr eines Kehlkopfkollapses besteht. Zudem ist die Schleimhaut im Bereich der Processus corniculates meist im Übermaß ausgebildet und geschwollen, so dass sie bei der Einatmung durch die Stimmritze gezogen wird und sie teilweise verlegt.
Im Bereich der Luftröhre und großen Bronchien treten rassespezifische Fehlbildungen auf. Auch die Knorpelspangen der Luftröhre sind beim Mops weich (Tracheomalazie), so dass sie zum Trachealkollaps neigen. Dagegen sind die Knorpelspangen der Französischen Bulldogge zwar klein, aber zumeist ausreichend steif (hypoplastische Trachea). Diese Veränderungen setzen sich auch auf die großen Bronchien fort.
Klinisches Bild
Typisches Symptom des brachycephalen Syndroms ist eine geräuschvolle, in der Regel inspiratorisch betonte Atmung in Verbindung mit Zeichen von Atemnot. Bei der klinischen Untersuchung können als charakteristische Befunde verengte Nasenlöcher und Nasenhöhlen, ein verlängertes und verdicktes Gaumensegel, verkürzter Rachenraum sowie Veränderungen am Kehlkopf festgestellt werden. Darüber hinaus können die Rachenmandeln in den Innenraum der Atemwege gezogen werden, wenn der Unterdruck beim Einatmen zu groß wird. Dies kann zu Atemproblemen, Erstickungsanfällen, Ohnmacht, zumindest aber röchelnden Atemgeräuschen und Schnarchgeräuschen führen. Durch die verminderte Fähigkeit zum Hecheln reagieren brachycephale Hunde empfindlicher auf Hitze als ihre nicht deformierten Artgenossen.
Mögliche Komplikationen eines brachycephalen Syndroms sind ein Ödem des Kehldeckels, Kollaps des Kehlkopfs, Einstülpung der seitlichen Kehlkopftaschen nach innen, Trachealkollaps, Entzündung und/oder Vorfall der Rachenmandeln, Bronchitis sowie Herzinsuffizienz infolge von ungenügender Sauerstoffsättigung des Blutes.
Häufig ist auch eine erhöhte Anfälligkeit gegenüber Wärme zu beobachten. Da die Nasenmuscheln und die laterale Nasendrüse eine wichtige Rolle bei der Wärmeabgabe spielen, sind brachycephale Hunde häufig sehr empfindlich gegenüber warmen Umgebungstemperaturen.
Therapie
Die Behandlung eines brachycephalen Syndroms kann über konservative oder chirurgische Maßnahmen erfolgen, wobei eine Gewichtsreduktion und konservative Behandlung mit Corticosteroiden nur in leichten Fällen Aussicht auf Erfolg bietet.
Die chirurgische Behandlung besteht in der Resektion des die Atemwege einengenden Gewebes (Nasenlocherweiterung, Vestibuloplastie, Laser-assistierte Turbinektomie, Gaumensegelresektion, Tonsillektomie etc.), um dadurch ein freieres Atmen zu ermöglichen.
Sonstige gesundheitliche Probleme
Die proportionale Vergrößerung des Kopfes in Verbindung mit der runden Kopfform führt mechanisch zu einem erhöhten Risiko für Schwergeburten. Auch bei einem Kaiserschnitt ist die Überlebensrate von brachycephalen Welpen im Vergleich zu anatomisch normalen Hunden verringert. Brachycephale Rassen sind außerdem überdurchschnittlich häufig von Hirntumoren und Wasserkopf betroffen. Besonders ausgeprägt ist diese Prädisposition bei Rassen, in denen zusätzlich zur Brachycephalie auch eine Chondrodysplasie vorhanden ist.
Die starke Abweichung von der normalen Kopfform führt außerdem zu einer anatomischen Reorganisation des Gehirns. Brachycephale Zwergrassen sind daneben häufig von bis ins Erwachsenenalter persistierenden Fontanellen betroffen, was durch den mangelhaften Schutz des Gehirns in der Schädelhöhle ein zusätzliches Verletzungsrisiko darstellt.
Brachycephalie führt durch die Verkürzung des Oberkiefers (Brachygnathia superior) oft zu einem ausgeprägten Vorbiss, der bei einigen brachycephalen Rassen auch ausdrücklich im Standard gefordert wird. Dies kann in manchen Fällen zu einer mangelhaften Gebissfunktion führen.
Bei den extrem rundköpfigen Rassen (z.B.Mops) sind zusätzlich hervorstehende, teils auch vergrößerte Augen zu beobachten, was zu häufigen Verletzungen der Hornhaut führt. Außerdem besteht die Gefahr eines Augapfelvorfalls. Dieser Symptomkomplex wird vereinzelt als okulares Brachycephalensyndrom bezeichnet.
Genetik und Zuchthygiene
Brachycephalie ist ein Komplex aus verschiedenen anatomischen Merkmalen, die polygen vererbt werden. Aus tierschützerischen Überlegungen sind extrem brachycephale Tiere von der Zucht auszuschließen.
Insbesondere ist die extreme Rundköpfigkeit in Kombination mit einer ausgeprägten Verkürzung der Gesichtsknochen züchterisch zu bekämpfen, was über das Festlegen von Grenzwerten und einer darauf basierenden Indexselektion und Zuchtwertschätzung möglich ist.
Zuchtversuche zur Bekämpfung von Brachycephalie und brachycephalem Syndrom sind beispielsweise der Continental Bulldog und die Olde English Bulldogge.
Betroffene Rassen
Folgende Rassen sind vom Auftreten von Brachycephalie und den Folgen aufgrund der Zuchtentwicklung der letzten Jahrzehnte betroffen:
- Malteser
- Mops
- Englische Bulldogge
- Französische Bulldogge
- Boston Terrier
- Boxer
- Shih-Tzu
- Pekinese
- Chihuahua
- King-Charles-Spaniel
- Belgische Zwerggriffons (Griffon Belge, Griffon Bruxellois, Petit Brabançon)
- Yorkshire-Terrier
- Zwergpinscher
Eine dem brachycephalen Syndrom ähnliche Erkrankung bei einer nicht eindeutig brachycephalen Hunderasse ist beim Norwich Terrier beschrieben, dessen Schädel eine nur bedingt brachycephale Anatomie aufweist.
Gesetzliche Situation
Starke Erscheinungsformen von Brachycephalie gelten laut einem Gutachten des Bundesministeriums für Verbraucherschutz, Ernährung und Landwirtschaft als verbotene Qualzucht. In dem Gutachten werden den Zuchtverbänden Untersuchungen betroffener Tiere, Zuchteinschränkungen und geänderte Zuchtstandards empfohlen.
In §11b des Tierschutzgesetzes, der das Verbot von Qualzucht regelt, ist festgelegt:
„Es ist verboten, Wirbeltiere zu züchten […], wenn damit gerechnet werden muss, dass bei der Nachzucht, […] erblich bedingt Körperteile oder Organe für den artgemäßen Gebrauch fehlen oder untauglich oder umgestaltet sind und hierdurch Schmerzen, Leiden oder Schäden auftreten.“
Die Definition eines Schädels als brachycephal geschieht nach mehreren Kriterien:
- Kurzer, breiter Gesichtsschädel
- Verhältnis Schädelbreite zu Schädellänge von 0,81 oder höher
- Verhältnis Hirnschädel zu Gesichtsschädel größer 1,6
- Kraniofazialwinkel zwischen 9° und 14°
Der Kraniofazialwinkel ist der von der Schnauzenspitze, der Schädelbasis und dem Mittelpunkt der Augenhöhle begrenzte Winkel.
Gesundheitliche Folgen
Brachycephales Syndrom
Brachycephalie kann zu Problemen der oberen Atemwege führen, die zusammenfassend als brachycephales Syndrom oder Brachycephalie-Syndrom bezeichnet werden. Es ist durch eine starke Behinderung der Atmung und eine gestörte Thermoregulation gekennzeichnet.
Pathogenese
Auslöser der Verengung der oberen Luftwege ist vor allem eine Hemmung des Längenwachstums des Gesichtsschädels. Durch das voluminöse Ausmaß des Knorpels des Nasenflügels (insbesondere die Cartilago nasi lateralis dorsalis) und einer reduzierten Beweglichkeit des Nasenflügels kommt es zu einer Verengung der Nasenlöcher.
Die Nasenhöhle des heranwachsenden Hundes streckt sich normalerweise in die Länge und die zur Geburt noch unterentwickelten Nasenmuscheln wachsen aufeinander zu, stellen aber ihr Wachstum ein bevor die sie überziehende Schleimhautschichten einander berühren. Bei brachycephalen Hunden unterbleibt diese Wachstumshemmung – es kommt zu einer relativen Conchen-Hypertrophie („Nasenmuschelwucherung“). Zudem wachsen die Nasenmuscheln als aberrante Nasenmuscheln über ein für die Größe der Nasenhöhle mögliches Maß.
Man unterscheidet dabei rostrale aberrante Nasenmuscheln (vor der Plica alaris gelegen), die vorderen Abschnitte des mittleren und unteren Nasengangs einengen, und kaudale aberrante Nasenmuscheln, die den Übergang zum Nasenrachen (Meatus nasopharyngeus) einengen. Zudem sind die Lamellen der Nasenmuscheln bei brachycephalen Hunden deutlich dicker als bei anderen Hunden. Die Folge ist, dass kaum noch Luft durch die Nase strömen kann, was für einen obligaten Nasenatmer ein schwerwiegendes Problem darstellt.
Das Gaumensegel brachycephaler Hunde ist verlängert und verdickt. Dies führt zu einer Einengung des Nasenrachens sowie in Kombination mit der relativ zu großen Zunge (relative Makroglossie) auch des Mundrachens.
Im Bereich des Kehlkopfs treten ebenfalls krankhafte Veränderungen auf, vor allem beim Mops. Hier ist das Kehlkopfskelett instabil, insbesondere der Stellknorpel und der Kehldeckel, so dass die Gefahr eines Kehlkopfkollapses besteht. Zudem ist die Schleimhaut im Bereich der Processus corniculates meist im Übermaß ausgebildet und geschwollen, so dass sie bei der Einatmung durch die Stimmritze gezogen wird und sie teilweise verlegt.
Im Bereich der Luftröhre und großen Bronchien treten rassespezifische Fehlbildungen auf. Auch die Knorpelspangen der Luftröhre sind beim Mops weich (Tracheomalazie), so dass sie zum Trachealkollaps neigen. Dagegen sind die Knorpelspangen der Französischen Bulldogge zwar klein, aber zumeist ausreichend steif (hypoplastische Trachea). Diese Veränderungen setzen sich auch auf die großen Bronchien fort.
Klinisches Bild
Typisches Symptom des brachycephalen Syndroms ist eine geräuschvolle, in der Regel inspiratorisch betonte Atmung in Verbindung mit Zeichen von Atemnot. Bei der klinischen Untersuchung können als charakteristische Befunde verengte Nasenlöcher und Nasenhöhlen, ein verlängertes und verdicktes Gaumensegel, verkürzter Rachenraum sowie Veränderungen am Kehlkopf festgestellt werden. Darüber hinaus können die Rachenmandeln in den Innenraum der Atemwege gezogen werden, wenn der Unterdruck beim Einatmen zu groß wird. Dies kann zu Atemproblemen, Erstickungsanfällen, Ohnmacht, zumindest aber röchelnden Atemgeräuschen und Schnarchgeräuschen führen. Durch die verminderte Fähigkeit zum Hecheln reagieren brachycephale Hunde empfindlicher auf Hitze als ihre nicht deformierten Artgenossen.
Mögliche Komplikationen eines brachycephalen Syndroms sind ein Ödem des Kehldeckels, Kollaps des Kehlkopfs, Einstülpung der seitlichen Kehlkopftaschen nach innen, Trachealkollaps, Entzündung und/oder Vorfall der Rachenmandeln, Bronchitis sowie Herzinsuffizienz infolge von ungenügender Sauerstoffsättigung des Blutes.
Häufig ist auch eine erhöhte Anfälligkeit gegenüber Wärme zu beobachten. Da die Nasenmuscheln und die laterale Nasendrüse eine wichtige Rolle bei der Wärmeabgabe spielen, sind brachycephale Hunde häufig sehr empfindlich gegenüber warmen Umgebungstemperaturen.
Therapie
Die Behandlung eines brachycephalen Syndroms kann über konservative oder chirurgische Maßnahmen erfolgen, wobei eine Gewichtsreduktion und konservative Behandlung mit Corticosteroiden nur in leichten Fällen Aussicht auf Erfolg bietet.
Die chirurgische Behandlung besteht in der Resektion des die Atemwege einengenden Gewebes (Nasenlocherweiterung, Vestibuloplastie, Laser-assistierte Turbinektomie, Gaumensegelresektion, Tonsillektomie etc.), um dadurch ein freieres Atmen zu ermöglichen.
Sonstige gesundheitliche Probleme
Die proportionale Vergrößerung des Kopfes in Verbindung mit der runden Kopfform führt mechanisch zu einem erhöhten Risiko für Schwergeburten. Auch bei einem Kaiserschnitt ist die Überlebensrate von brachycephalen Welpen im Vergleich zu anatomisch normalen Hunden verringert. Brachycephale Rassen sind außerdem überdurchschnittlich häufig von Hirntumoren und Wasserkopf betroffen. Besonders ausgeprägt ist diese Prädisposition bei Rassen, in denen zusätzlich zur Brachycephalie auch eine Chondrodysplasie vorhanden ist.
Die starke Abweichung von der normalen Kopfform führt außerdem zu einer anatomischen Reorganisation des Gehirns. Brachycephale Zwergrassen sind daneben häufig von bis ins Erwachsenenalter persistierenden Fontanellen betroffen, was durch den mangelhaften Schutz des Gehirns in der Schädelhöhle ein zusätzliches Verletzungsrisiko darstellt.
Brachycephalie führt durch die Verkürzung des Oberkiefers (Brachygnathia superior) oft zu einem ausgeprägten Vorbiss, der bei einigen brachycephalen Rassen auch ausdrücklich im Standard gefordert wird. Dies kann in manchen Fällen zu einer mangelhaften Gebissfunktion führen.
Bei den extrem rundköpfigen Rassen (z.B.Mops) sind zusätzlich hervorstehende, teils auch vergrößerte Augen zu beobachten, was zu häufigen Verletzungen der Hornhaut führt. Außerdem besteht die Gefahr eines Augapfelvorfalls. Dieser Symptomkomplex wird vereinzelt als okulares Brachycephalensyndrom bezeichnet.
Genetik und Zuchthygiene
Brachycephalie ist ein Komplex aus verschiedenen anatomischen Merkmalen, die polygen vererbt werden. Aus tierschützerischen Überlegungen sind extrem brachycephale Tiere von der Zucht auszuschließen.
Insbesondere ist die extreme Rundköpfigkeit in Kombination mit einer ausgeprägten Verkürzung der Gesichtsknochen züchterisch zu bekämpfen, was über das Festlegen von Grenzwerten und einer darauf basierenden Indexselektion und Zuchtwertschätzung möglich ist.
Zuchtversuche zur Bekämpfung von Brachycephalie und brachycephalem Syndrom sind beispielsweise der Continental Bulldog und die Olde English Bulldogge.
Betroffene Rassen
Folgende Rassen sind vom Auftreten von Brachycephalie und den Folgen aufgrund der Zuchtentwicklung der letzten Jahrzehnte betroffen:
- Malteser
- Mops
- Englische Bulldogge
- Französische Bulldogge
- Boston Terrier
- Boxer
- Shih-Tzu
- Pekinese
- Chihuahua
- King-Charles-Spaniel
- Belgische Zwerggriffons (Griffon Belge, Griffon Bruxellois, Petit Brabançon)
- Yorkshire-Terrier
- Zwergpinscher
Eine dem brachycephalen Syndrom ähnliche Erkrankung bei einer nicht eindeutig brachycephalen Hunderasse ist beim Norwich Terrier beschrieben, dessen Schädel eine nur bedingt brachycephale Anatomie aufweist.
Gesetzliche Situation
Starke Erscheinungsformen von Brachycephalie gelten laut einem Gutachten des Bundesministeriums für Verbraucherschutz, Ernährung und Landwirtschaft als verbotene Qualzucht. In dem Gutachten werden den Zuchtverbänden Untersuchungen betroffener Tiere, Zuchteinschränkungen und geänderte Zuchtstandards empfohlen.
In §11b des Tierschutzgesetzes, der das Verbot von Qualzucht regelt, ist festgelegt:
„Es ist verboten, Wirbeltiere zu züchten […], wenn damit gerechnet werden muss, dass bei der Nachzucht, […] erblich bedingt Körperteile oder Organe für den artgemäßen Gebrauch fehlen oder untauglich oder umgestaltet sind und hierdurch Schmerzen, Leiden oder Schäden auftreten.“
eingestellt von: ise; Quellen- und Bildnachweis

Canine Ceroid-Lipofuszinose (CCL)
Die Canine Ceroid-Lipofuszinose (CCL) ist eine Erbkrankheit bei verschiedenen Hunderassen, welche Körperzellen, insbesondere Nervenzellen schädigt. Sie entspricht der Neuronalen Ceroid-Lipofuszinose (NCL) des Menschen und wird in der Literatur auch als NCL der Hunde bezeichnet. CCL ist unheilbar und verläuft immer tödlich.
Pathophysiologie
Bei der CCL handelt es sich um eine Gruppe von Lysosomalen Speicherkrankheiten, die je nach Hunderasse auf verschiedenen Mutationen beruht. Sie werden alle einfach autosomal rezessiv vererbt und führen zur kontinuierlichen Einlagerung vom Ceroid und Lipofuszin in den Nervenzellen, wobei das Alter bei der Erstdiagnose und die durchschnittliche Lebenserwartung in Abhängigkeit von der auslösenden Mutation variieren können.
Durch die Einlagerung der Substanzen wird die Funktion der Nervenzellen beeinträchtigt, was zu einer fortschreitenden Degeneration des Nervensystems und entsprechenden neurologischen Symptomen führt.
Klinik
Signalement
Das Alter, in dem die Symptome der Erkrankung zuerst auffällig werden, variiert je nach Rasse zwischen etwa sechs Monaten und sechs bis sieben Jahren.
Hunde der Rassen Dalmatiner und Australian Cattle Dog werden normalerweise bereits im ersten Lebensjahr vorgestellt; Chihuahuas, Zwergschnauzer und Dachsbracken zwischen zwei und vier Jahren; Labrador Retriever und Welsh Corgi normalerweise erst ab sechs Jahren. Bei English Cocker Spaniels (1.5-6) und Polski Owczarek Nizinny (0.5-4.5) variiert das Diagnosealter stark. Beim Dackel kommen zwei Formen der Erkrankung vor, die etwa im Alter von neun Monaten bzw. zwischen viereinhalb und sechseinhalb Jahren auftreten.
Symptome
Die Symptome ergeben sich aus dem Funktionsverlust des Nervensystems, der durch die Einlagerung von Ceroid und Lipofuszin entsteht. Es kommt dabei zu funktionellen Ausfällen wie Ataxie, aber auch zu Veränderungen der Persönlichkeit wie etwa unsicherem Verhalten in vertrauter Umgebung, Demenz, Aggressivität, Desorientierung und Verlust der Stubenreinheit. Die Symptome verschlimmern sich mit der Zeit.
Genetik und Zuchthygiene
Bisher sind alle bekannten Varianten der CCL als einfach autosomal rezessiv beschrieben.
Daraus ergibt sich, dass die Elterntiere eines betroffenen Hundes sowie alle seine bereits geborenen Nachkommen der 1. Generation sicher Träger sind und zwei Drittel der klinisch gesunden Vollgeschwister befallener Welpen ebenfalls das defekte Allel tragen; sie sind somit von der Zucht auszuschließen.
Bei Rassen, für die ein Gentest vorliegt (American Bulldog, Border Collie, English Setter, Dackel (Gentest nur für die CCL beim Junghund), Tibet-Terrier), können Träger des defekten Allels identifiziert werden.
Indem solche Träger nur mit Hunden verpaart werden, bei denen ein Gentest gezeigt hat, dass sie zwei gesunde Allele tragen, kann das klinische Auftreten der Krankheit in der Population vermieden werden.
Pathophysiologie
Bei der CCL handelt es sich um eine Gruppe von Lysosomalen Speicherkrankheiten, die je nach Hunderasse auf verschiedenen Mutationen beruht. Sie werden alle einfach autosomal rezessiv vererbt und führen zur kontinuierlichen Einlagerung vom Ceroid und Lipofuszin in den Nervenzellen, wobei das Alter bei der Erstdiagnose und die durchschnittliche Lebenserwartung in Abhängigkeit von der auslösenden Mutation variieren können.
Durch die Einlagerung der Substanzen wird die Funktion der Nervenzellen beeinträchtigt, was zu einer fortschreitenden Degeneration des Nervensystems und entsprechenden neurologischen Symptomen führt.
Klinik
Signalement
Das Alter, in dem die Symptome der Erkrankung zuerst auffällig werden, variiert je nach Rasse zwischen etwa sechs Monaten und sechs bis sieben Jahren.
Hunde der Rassen Dalmatiner und Australian Cattle Dog werden normalerweise bereits im ersten Lebensjahr vorgestellt; Chihuahuas, Zwergschnauzer und Dachsbracken zwischen zwei und vier Jahren; Labrador Retriever und Welsh Corgi normalerweise erst ab sechs Jahren. Bei English Cocker Spaniels (1.5-6) und Polski Owczarek Nizinny (0.5-4.5) variiert das Diagnosealter stark. Beim Dackel kommen zwei Formen der Erkrankung vor, die etwa im Alter von neun Monaten bzw. zwischen viereinhalb und sechseinhalb Jahren auftreten.
Symptome
Die Symptome ergeben sich aus dem Funktionsverlust des Nervensystems, der durch die Einlagerung von Ceroid und Lipofuszin entsteht. Es kommt dabei zu funktionellen Ausfällen wie Ataxie, aber auch zu Veränderungen der Persönlichkeit wie etwa unsicherem Verhalten in vertrauter Umgebung, Demenz, Aggressivität, Desorientierung und Verlust der Stubenreinheit. Die Symptome verschlimmern sich mit der Zeit.
Genetik und Zuchthygiene
Bisher sind alle bekannten Varianten der CCL als einfach autosomal rezessiv beschrieben.
Daraus ergibt sich, dass die Elterntiere eines betroffenen Hundes sowie alle seine bereits geborenen Nachkommen der 1. Generation sicher Träger sind und zwei Drittel der klinisch gesunden Vollgeschwister befallener Welpen ebenfalls das defekte Allel tragen; sie sind somit von der Zucht auszuschließen.
Bei Rassen, für die ein Gentest vorliegt (American Bulldog, Border Collie, English Setter, Dackel (Gentest nur für die CCL beim Junghund), Tibet-Terrier), können Träger des defekten Allels identifiziert werden.
Indem solche Träger nur mit Hunden verpaart werden, bei denen ein Gentest gezeigt hat, dass sie zwei gesunde Allele tragen, kann das klinische Auftreten der Krankheit in der Population vermieden werden.
eingestellt von: ise; Quellennachweis
Collie Eye Anomaly

Die Collie Eye Anomaly (dt. Collieaugen-Anomalie, abgekürzt CEA) ist eine Erbkrankheit verschiedener Hunderassen aus der Familie der Collies und der mit ihnen verwandten Rassen.
Die Krankheit betrifft den Augenhintergrund und führt zu einer angeborenen Beeinträchtigung der Sehkraft bis hin zur Erblindung.
Pathophysiologie
Die Collie Eye Anomaly beruht auf einer genetisch bedingten Fehlbildung und Hypoplasie der Aderhaut und Netzhaut im Laufe der Embryonalentwicklung. Dadurch kommt es zu Störungen in der Entwicklung der Blutgefäße, die zu Blutungen ins Auge führen können.
Ebenfalls können Kolobome der Ader- und Netzhaut auftreten. Auch eine Netzhautablösung ist möglich. Je nach Schweregrad der Veränderungen kann die Sehkraft nicht beeinträchtigt sein; schwerere Fälle können aber eine verminderte Sehkraft bis hin zu völliger Erblindung zeigen.
Klinik
Signalement
Die Krankheit tritt bei Langhaar- und Kurzhaarcollies sowie Shelties und verwandten Rassen wie Border Collie, Australian Shepherd und auch Lancashire Heeler auf. Schwere Fälle werden normalerweise bereits im Welpenalter diagnostiziert; leichtere Fälle können zu jedem Lebenszeitpunkt als Zufallsdiagnose festgestellt werden.
Symptome
Betroffene Welpen zeigen von Anfang an eine verringerte Sehkraft bis hin zur Erblindung. Die Erkrankung schreitet normalerweise nicht fort; allenfalls können schwerere Kolobome später zu einer Netzhautablösung führen. Leichtere Fälle sind klinisch normal und können nur mittels Ophthalmoskopie diagnostiziert werden.
Diagnose
Die Diagnose erfolgt mittels Ophthalmoskopie, mittels derer die charakteristischen Veränderungen im Augenhintergrund festgestellt werden können. Bei erkrankten Hunden sind normalerweise beide Augen betroffen; allerdings kann der Schweregrad der Erkrankung zwischen den beiden Augen unterschiedlich sein.
Therapie und Prognose
Eine Heilung ist nicht möglich. Netzhautablösungen können in manchen Fällen laserchirurgisch behandelt werden. Ein Fortschreiten der Erkrankung ist bei Hunden mit geringgradiger CEA nicht zu erwarten. Unterstützende Maßnahmen beschränken sich darauf, die Umwelt des Hundes an seine verringerte Sehkraft anzupassen und ihm dadurch eine gute Lebensqualität zu ermöglichen. Die Lebenserwartung ist im Vergleich zu gesunden Hunden nicht verringert.
Genetik und Zuchthygiene
Die Collie Eye Anomaly scheint durch mehrere Loci kontrolliert zu werden (polygener Erbgang). 80 bis 90 Prozent der Collies weisen Veränderungen des Augenhintergrunds auf, ohne dass ihre Sehkraft eingeschränkt ist.
Auch Collies ohne Veränderungen des Augenhintergrunds können Träger der Erkrankung sein. Es wird empfohlen, alle Welpen gefährdeter Rassen ophthalmoskopisch zu screenen und die so erhaltenen Daten mittels einer zentralen Datenbank für die Zuchtwertschätzung zu verwenden.
Mittels Screening und Zuchteinschränkungen kann die Anzahl betroffener Tiere signifikant reduziert werden. Bei vielen Rassen weisen betroffene Hunde eine 7.8 kb lange Deletion im NHEJ1-Gen auf. Ein Gentest ist inzwischen ebenfalls verfügbar.
Die Krankheit betrifft den Augenhintergrund und führt zu einer angeborenen Beeinträchtigung der Sehkraft bis hin zur Erblindung.
Pathophysiologie
Die Collie Eye Anomaly beruht auf einer genetisch bedingten Fehlbildung und Hypoplasie der Aderhaut und Netzhaut im Laufe der Embryonalentwicklung. Dadurch kommt es zu Störungen in der Entwicklung der Blutgefäße, die zu Blutungen ins Auge führen können.
Ebenfalls können Kolobome der Ader- und Netzhaut auftreten. Auch eine Netzhautablösung ist möglich. Je nach Schweregrad der Veränderungen kann die Sehkraft nicht beeinträchtigt sein; schwerere Fälle können aber eine verminderte Sehkraft bis hin zu völliger Erblindung zeigen.
Klinik
Signalement
Die Krankheit tritt bei Langhaar- und Kurzhaarcollies sowie Shelties und verwandten Rassen wie Border Collie, Australian Shepherd und auch Lancashire Heeler auf. Schwere Fälle werden normalerweise bereits im Welpenalter diagnostiziert; leichtere Fälle können zu jedem Lebenszeitpunkt als Zufallsdiagnose festgestellt werden.
Symptome
Betroffene Welpen zeigen von Anfang an eine verringerte Sehkraft bis hin zur Erblindung. Die Erkrankung schreitet normalerweise nicht fort; allenfalls können schwerere Kolobome später zu einer Netzhautablösung führen. Leichtere Fälle sind klinisch normal und können nur mittels Ophthalmoskopie diagnostiziert werden.
Diagnose
Die Diagnose erfolgt mittels Ophthalmoskopie, mittels derer die charakteristischen Veränderungen im Augenhintergrund festgestellt werden können. Bei erkrankten Hunden sind normalerweise beide Augen betroffen; allerdings kann der Schweregrad der Erkrankung zwischen den beiden Augen unterschiedlich sein.
Therapie und Prognose
Eine Heilung ist nicht möglich. Netzhautablösungen können in manchen Fällen laserchirurgisch behandelt werden. Ein Fortschreiten der Erkrankung ist bei Hunden mit geringgradiger CEA nicht zu erwarten. Unterstützende Maßnahmen beschränken sich darauf, die Umwelt des Hundes an seine verringerte Sehkraft anzupassen und ihm dadurch eine gute Lebensqualität zu ermöglichen. Die Lebenserwartung ist im Vergleich zu gesunden Hunden nicht verringert.
Genetik und Zuchthygiene
Die Collie Eye Anomaly scheint durch mehrere Loci kontrolliert zu werden (polygener Erbgang). 80 bis 90 Prozent der Collies weisen Veränderungen des Augenhintergrunds auf, ohne dass ihre Sehkraft eingeschränkt ist.
Auch Collies ohne Veränderungen des Augenhintergrunds können Träger der Erkrankung sein. Es wird empfohlen, alle Welpen gefährdeter Rassen ophthalmoskopisch zu screenen und die so erhaltenen Daten mittels einer zentralen Datenbank für die Zuchtwertschätzung zu verwenden.
Mittels Screening und Zuchteinschränkungen kann die Anzahl betroffener Tiere signifikant reduziert werden. Bei vielen Rassen weisen betroffene Hunde eine 7.8 kb lange Deletion im NHEJ1-Gen auf. Ein Gentest ist inzwischen ebenfalls verfügbar.
eingestellt von: ise; Quellen- und Bildnachweis

Dackellähme
Als Dackellähme (oder auch Teckellähme) werden die durch einen Bandscheibenvorfall ausgelösten Krankheitssymptome bei Hunden, welche eine genetisch fixierte Knorpelwachstumsstörung (Chondrodystrophie) aufweisen, bezeichnet.
Wie der Name bereits sagt, sind hiervon häufig Dackel betroffen, aber auch Pekinesen, Scottish Terrier, Spaniel, Französische Bulldoggen und Zwergpudel sind für die Erkrankung anfällig.
Pathogenese
Die Erkrankung beruht auf einer Degeneration der Bandscheiben.
Diese besteht aus dem peripher liegenden Anulus fibrosus und dem zentral liegenden Bandscheibenkern (Nucleus pulposus). Die vorliegende Chondrodystrophie führt zu einer Umwandlung des elastischen, gallertigen Kernes in knorpelartiges, verkalktes, teilweise abgestorbenes Gewebe, der mit einer zunehmenden Instabilität und Auffaserung des Anulis fibrosus einhergeht (Enchondrosis intervertebralis). Die somit kaum noch elastische und belastbare Bandscheibe kann somit bereits bei kleineren Belastungen oder Traumen reißen: es kommt zum Vorfall des Bandscheibenkerns oder der gesamten Bandscheibe in den Wirbelkanal hinein.
Hierdurch werden Quetschungen und Schädigungen des Nervengewebes verursacht, welche wiederum entsprechende klinische Ausfallserscheinungen der Nervenfunktion nach sich zieht. Die Erkrankung tritt vor allem im jüngeren bis mittleren Lebensalter im Alter zwischen zwei und sieben Jahren auf und betrifft vor allem den mechanisch am stärksten beanspruchten Teil der Wirbelsäule zwischen elftem Brust- und drittem Lendenwirbel, seltener die kaudale Lendenwirbelsäule zwischen fünftem Lendenwirbel und Kreuzbein sowie die Halswirbelsäule zwischen dem zweiten und siebten Halswirbel.
Klinik
Die Symptomatik ist durch eine deutliche Bewegungsstörung der betroffenen Tiere gekennzeichnet. Diese ist abhängig von der Schwere des Vorfalls auf Schmerzhaftigkeit im Bereich des Rückens oder auf den mehr oder weniger starken Ausfall von Nervenfunktionen zurückzuführen.
Im Extremfall kommt es zu einer kompletten Lähmung der Beckengliedmaßen mit spastischer Parese oder schlaffer Lähmung. Die Fähigkeit zu Harn- und Kotabsatz ist häufig eingeschränkt. Bei einem Vorfall im Bereich der Halswirbelsäule können analog die Vordergliedmaßen zusätzlich betroffen sein. Bei länger bestehender Symptomatik sind Hautschäden durch Druckstellen möglich.
Diagnose
Gemeinsam mit der Rasse des betroffenen Tieres gibt eine neurologische Untersuchung deutliche Anhaltspunkte für die Lokalisation der Schädigung. Dagegen ist das Anfertigen einer einfachen Röntgenaufnahme nicht immer hilfreich, da sich die vorgefallenen Bandscheibenanteile meist im Röntgen nicht deutlich nachweisen lassen. Mittels einer Myelographie ist die Eingrenzung des Schadens jedoch in der Regel möglich.
Letzte Sicherheit über das Ausmaß des Schadens kann eine Magnetresonanztomographie geben.
Therapie
Liegt keine hochgradige Schädigung vor, ist eine konservative Therapie mittels Medikamenten (Glukokortikoide, Antiphlogistika, Anabolika und Vitamin-B-Komplex) möglich und zielt auf das Zurückdrängen entzündlicher Reaktionen ab.
Die chirurgische Intervention ist dagegen auf die Druckentlastung des Rückenmarks ausgerichtet, was über Fensterung (Fenestrierung) der Bandscheibe sowie über Entfernung von Teilen des angrenzenden Wirbeldaches (Laminektomie oder Hemilaminektomie) erfolgen kann.
Nach erfolgter Operation kann mittels Physiotherapie die Genesung beschleunigt werden
Wie der Name bereits sagt, sind hiervon häufig Dackel betroffen, aber auch Pekinesen, Scottish Terrier, Spaniel, Französische Bulldoggen und Zwergpudel sind für die Erkrankung anfällig.
Pathogenese
Die Erkrankung beruht auf einer Degeneration der Bandscheiben.
Diese besteht aus dem peripher liegenden Anulus fibrosus und dem zentral liegenden Bandscheibenkern (Nucleus pulposus). Die vorliegende Chondrodystrophie führt zu einer Umwandlung des elastischen, gallertigen Kernes in knorpelartiges, verkalktes, teilweise abgestorbenes Gewebe, der mit einer zunehmenden Instabilität und Auffaserung des Anulis fibrosus einhergeht (Enchondrosis intervertebralis). Die somit kaum noch elastische und belastbare Bandscheibe kann somit bereits bei kleineren Belastungen oder Traumen reißen: es kommt zum Vorfall des Bandscheibenkerns oder der gesamten Bandscheibe in den Wirbelkanal hinein.
Hierdurch werden Quetschungen und Schädigungen des Nervengewebes verursacht, welche wiederum entsprechende klinische Ausfallserscheinungen der Nervenfunktion nach sich zieht. Die Erkrankung tritt vor allem im jüngeren bis mittleren Lebensalter im Alter zwischen zwei und sieben Jahren auf und betrifft vor allem den mechanisch am stärksten beanspruchten Teil der Wirbelsäule zwischen elftem Brust- und drittem Lendenwirbel, seltener die kaudale Lendenwirbelsäule zwischen fünftem Lendenwirbel und Kreuzbein sowie die Halswirbelsäule zwischen dem zweiten und siebten Halswirbel.
Klinik
Die Symptomatik ist durch eine deutliche Bewegungsstörung der betroffenen Tiere gekennzeichnet. Diese ist abhängig von der Schwere des Vorfalls auf Schmerzhaftigkeit im Bereich des Rückens oder auf den mehr oder weniger starken Ausfall von Nervenfunktionen zurückzuführen.
Im Extremfall kommt es zu einer kompletten Lähmung der Beckengliedmaßen mit spastischer Parese oder schlaffer Lähmung. Die Fähigkeit zu Harn- und Kotabsatz ist häufig eingeschränkt. Bei einem Vorfall im Bereich der Halswirbelsäule können analog die Vordergliedmaßen zusätzlich betroffen sein. Bei länger bestehender Symptomatik sind Hautschäden durch Druckstellen möglich.
Diagnose
Gemeinsam mit der Rasse des betroffenen Tieres gibt eine neurologische Untersuchung deutliche Anhaltspunkte für die Lokalisation der Schädigung. Dagegen ist das Anfertigen einer einfachen Röntgenaufnahme nicht immer hilfreich, da sich die vorgefallenen Bandscheibenanteile meist im Röntgen nicht deutlich nachweisen lassen. Mittels einer Myelographie ist die Eingrenzung des Schadens jedoch in der Regel möglich.
Letzte Sicherheit über das Ausmaß des Schadens kann eine Magnetresonanztomographie geben.
Therapie
Liegt keine hochgradige Schädigung vor, ist eine konservative Therapie mittels Medikamenten (Glukokortikoide, Antiphlogistika, Anabolika und Vitamin-B-Komplex) möglich und zielt auf das Zurückdrängen entzündlicher Reaktionen ab.
Die chirurgische Intervention ist dagegen auf die Druckentlastung des Rückenmarks ausgerichtet, was über Fensterung (Fenestrierung) der Bandscheibe sowie über Entfernung von Teilen des angrenzenden Wirbeldaches (Laminektomie oder Hemilaminektomie) erfolgen kann.
Nach erfolgter Operation kann mittels Physiotherapie die Genesung beschleunigt werden
eingestellt von: ise; Quellennachweis
Dalmatiner-Leukodystrophie
Die Dalmatiner-Leukodystrophie ist eine bei Dalmatinern vorkommende Erbkrankheit mit Entmarkung der weißen Substanz und Schwund des Großhirns mit Erweiterung der Hirnventrikel. Ab dem 3. Lebensmonat entwickeln sich Sehstörungen und Ataxie. Die Erkrankung führt schnell zum Verlust des Stehvermögens.
eingestellt von: ise; Quellennachweis
Dancing Dobermann Disease
Die Dancing Dobermann Disease (engl. für „Tanzender-Dobermann-Krankheit“) ist eine vermutlich degenerativ bedingte neurologische Erkrankung. Sie ist selten und tritt ausschließlich bei Dobermännern und anderen Pinschern auf.
Die Erkrankung tritt bei jungen ausgewachsenen Tieren erstmals auf und verläuft dann allmählich fortschreitend. Das Hauptsymptom ist eine Beugung des Sprunggelenks, zunächst meist einseitig. Diese kommt durch einen Ausfall des Musculus gastrocnemius und des Nervus tibialis zustande.
Im weiteren Verlauf kann es zur Lähmung beider Beine (Paraparese), Störung der Propriozeption, einem gesteigerten Patellarsehnenreflex und zu einer Atrophie des Musculus gastrocnemius kommen. Durch die zunehmende Schwäche in der Hinterhand entwickelt sich ein taumelnder, tänzelnder Gang, der der Krankheit ihren Namen gab.
Die Diagnose wird durch Vorbericht, klinisches Bild und eventuell Elektromyografie gestellt. Eine Heilung ist nicht möglich. Da die Erkrankung aber sehr langsam fortschreitet, kann über mehrere Jahre eine angemessene Lebensqualität aufrechterhalten werden.
Die Erkrankung tritt bei jungen ausgewachsenen Tieren erstmals auf und verläuft dann allmählich fortschreitend. Das Hauptsymptom ist eine Beugung des Sprunggelenks, zunächst meist einseitig. Diese kommt durch einen Ausfall des Musculus gastrocnemius und des Nervus tibialis zustande.
Im weiteren Verlauf kann es zur Lähmung beider Beine (Paraparese), Störung der Propriozeption, einem gesteigerten Patellarsehnenreflex und zu einer Atrophie des Musculus gastrocnemius kommen. Durch die zunehmende Schwäche in der Hinterhand entwickelt sich ein taumelnder, tänzelnder Gang, der der Krankheit ihren Namen gab.
Die Diagnose wird durch Vorbericht, klinisches Bild und eventuell Elektromyografie gestellt. Eine Heilung ist nicht möglich. Da die Erkrankung aber sehr langsam fortschreitet, kann über mehrere Jahre eine angemessene Lebensqualität aufrechterhalten werden.
eingestellt von: ise; Quellennachweis
Degenerative Myelopathie des Zwergpudels
Die degenerative Myelopathie des Zwergpudels ist eine vermutlich angeborene Demyelinisierung des Rückenmarks und Mittelhirns. Mit dem 2. bis 4. Lebensmonat entwickeln betroffene Zwergpudel zunehmend Paresen, die schließlich zu einer Tetraplegie führen können.
eingestellt von: ise; Quellennachweis
Degenerative Myelopathien der Hunde

Als Degenerative Myelopathien der Hunde fasst man eine Reihe langsam verlaufender neurologischer Erkrankungen zusammen, die mit einer Zerstörung des Rückenmarks einhergehen.
Diese Erkrankungen gehen mit langsam fortschreitenden Bewegungsstörungen der Hinterhand einher und sind nicht schmerzhaft. Eine Behandlung ist wenig erfolgversprechend. Die Degenerative Myelopathien der Hunde lassen sich nach der Altersverteilung in zwei große Gruppen einteilen, in die der alten Hunde und die der Junghunde.
Degenerative Myelopathie älterer Hunde
Die Degenerative Myelopathie älterer Hunde ist relativ häufig, besonders bekannt ist sie beim Deutschen Schäferhund. Sie entwickelt sich ab dem 5. Lebensjahr. Ursächlich ist eine Mutation des SOD1-Gens verantwortlich.Die Erkrankung ist durch eine Degeneration des Myelins im Brust- und Lendenteil des Rückenmarks gekennzeichnet. Dadurch entwickeln sich allmählich unkoordinierte Bewegungen der Hinterhand, eine gestörte Eigenwahrnehmung und gestörte Reflexe. Die Erkrankung ist nicht schmerzhaft.
Die Diagnose wird zumeist nach dem Ausschlussverfahren gestellt, sie kann nur nach Autopsie als sicher betrachtet werden. Vor allem ein Bandscheibenvorfall und eine Fibrokartilaginöse Embolie (beide treten akut auf), Cauda-equina-Syndrom und Wobbler-Syndrom (Röntgen, Myelografie) und schließlich Tumoren des Rückenmarks müssen ausgeschlossen werden. In der Rückenmarksflüssigkeit kann eine leichte Erhöhung des Proteingehalts auftreten. Eine Magnetresonanztomographie kann die Diagnose sichern, ist in der Tiermedizin aber nur selten verfügbar. Typisch und für die Abgrenzung zu anderen Erkrankungen wichtig, verläuft degenerative Myelopathie schmerzlos.
Mittels Physiotherapie, täglichem Laufen oder Schwimmen kann versucht werden, den Muskelschwund aufzuhalten. Behandlungsversuche wurden mit Prednisolon, Aminocapronsäure, N-Acetylcystein, Vitamin E, Vitamin C und Vitamin B-Komplex durchgeführt. Alle Medikationen zeigen nach einer aktuellen Studie keine erhöhte Wirksamkeit gegenüber der Physiotherapie und konnten den Krankheitsverlauf nicht aufhalten. Die Prognose ist daher schlecht und oftmals bleibt nur die Einschläferung, um dem erkrankten Tier unnötiges Leiden zu ersparen.
Degenerative Myelopathien der Junghunde
Im Gegensatz zur Degenerativen Myelopathie älterer Hunde sind die Degenerativen Myelopathien der Junghunde insgesamt sehr selten. Sie können allesamt nicht therapiert werden und haben eine schlechte Prognose. Differentialdiagnostisch müssen bei diesen Erkrankungen vor allem angeborene Fehlbildungen des Rückenmarks (Syringomyelie, Hydromyelie) und infektiöse Erkrankungen (Toxoplasmose, Neosporose, Staupe) berücksichtigt werden.
Die Spinale Muskelatrophie
(Stockard disease) ist eine degenerative Erkrankung der grauen Substanz, insbesondere der Motoneurone. Sie tritt bei Rottweiler, Deutscher Dogge, Dobermann, English Pointer, Epagneul Breton und Lapphund auf. Beim Epagneul Breton wurde ein autosomal-dominanter Erbgang nachgewiesen. Die Erkrankung beginnt im Welpenalter mit Atrophie der Rückenmuskulatur, später entwickeln sich Paresen oder sogar Paralysen. Bei Rottweilern kann sich ein Megaösophagus entwickeln (siehe auch Spinale Muskelatrophie des Menschen).
Die Progressive Degeneration des Ibiza-Hundes
ist eine mit den ersten Gehversuchen bei Ibiza-Welpen in Erscheinung tretende progressive Lähmung. Sie beginnt an der Hinterhand und greift dann auch auf die Vorderhand über. Spastizität und Dysmetrien kommen hinzu, gelegentlich werden auch Anfälle beobachtet.
Diese Erkrankungen gehen mit langsam fortschreitenden Bewegungsstörungen der Hinterhand einher und sind nicht schmerzhaft. Eine Behandlung ist wenig erfolgversprechend. Die Degenerative Myelopathien der Hunde lassen sich nach der Altersverteilung in zwei große Gruppen einteilen, in die der alten Hunde und die der Junghunde.
Degenerative Myelopathie älterer Hunde
Die Degenerative Myelopathie älterer Hunde ist relativ häufig, besonders bekannt ist sie beim Deutschen Schäferhund. Sie entwickelt sich ab dem 5. Lebensjahr. Ursächlich ist eine Mutation des SOD1-Gens verantwortlich.Die Erkrankung ist durch eine Degeneration des Myelins im Brust- und Lendenteil des Rückenmarks gekennzeichnet. Dadurch entwickeln sich allmählich unkoordinierte Bewegungen der Hinterhand, eine gestörte Eigenwahrnehmung und gestörte Reflexe. Die Erkrankung ist nicht schmerzhaft.
Die Diagnose wird zumeist nach dem Ausschlussverfahren gestellt, sie kann nur nach Autopsie als sicher betrachtet werden. Vor allem ein Bandscheibenvorfall und eine Fibrokartilaginöse Embolie (beide treten akut auf), Cauda-equina-Syndrom und Wobbler-Syndrom (Röntgen, Myelografie) und schließlich Tumoren des Rückenmarks müssen ausgeschlossen werden. In der Rückenmarksflüssigkeit kann eine leichte Erhöhung des Proteingehalts auftreten. Eine Magnetresonanztomographie kann die Diagnose sichern, ist in der Tiermedizin aber nur selten verfügbar. Typisch und für die Abgrenzung zu anderen Erkrankungen wichtig, verläuft degenerative Myelopathie schmerzlos.
Mittels Physiotherapie, täglichem Laufen oder Schwimmen kann versucht werden, den Muskelschwund aufzuhalten. Behandlungsversuche wurden mit Prednisolon, Aminocapronsäure, N-Acetylcystein, Vitamin E, Vitamin C und Vitamin B-Komplex durchgeführt. Alle Medikationen zeigen nach einer aktuellen Studie keine erhöhte Wirksamkeit gegenüber der Physiotherapie und konnten den Krankheitsverlauf nicht aufhalten. Die Prognose ist daher schlecht und oftmals bleibt nur die Einschläferung, um dem erkrankten Tier unnötiges Leiden zu ersparen.
Degenerative Myelopathien der Junghunde
Im Gegensatz zur Degenerativen Myelopathie älterer Hunde sind die Degenerativen Myelopathien der Junghunde insgesamt sehr selten. Sie können allesamt nicht therapiert werden und haben eine schlechte Prognose. Differentialdiagnostisch müssen bei diesen Erkrankungen vor allem angeborene Fehlbildungen des Rückenmarks (Syringomyelie, Hydromyelie) und infektiöse Erkrankungen (Toxoplasmose, Neosporose, Staupe) berücksichtigt werden.
Die Spinale Muskelatrophie
(Stockard disease) ist eine degenerative Erkrankung der grauen Substanz, insbesondere der Motoneurone. Sie tritt bei Rottweiler, Deutscher Dogge, Dobermann, English Pointer, Epagneul Breton und Lapphund auf. Beim Epagneul Breton wurde ein autosomal-dominanter Erbgang nachgewiesen. Die Erkrankung beginnt im Welpenalter mit Atrophie der Rückenmuskulatur, später entwickeln sich Paresen oder sogar Paralysen. Bei Rottweilern kann sich ein Megaösophagus entwickeln (siehe auch Spinale Muskelatrophie des Menschen).
Die Progressive Degeneration des Ibiza-Hundes
ist eine mit den ersten Gehversuchen bei Ibiza-Welpen in Erscheinung tretende progressive Lähmung. Sie beginnt an der Hinterhand und greift dann auch auf die Vorderhand über. Spastizität und Dysmetrien kommen hinzu, gelegentlich werden auch Anfälle beobachtet.
eingestellt von: ise; Quellen- und Bildnachweis
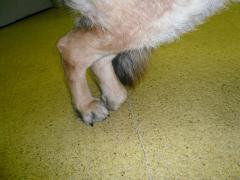
Ellbogendysplasie
Die Ellbogendysplasie (ED) ist ein chronisch verlaufender Krankheitskomplex des Ellbogengelenks schnellwüchsiger Hunderassen. Die ED stellt eine vererbte Entwicklungsstörung des wachsenden Skeletts dar.
Hohes Körpermassewachstum und Fütterungsfehler sind weitere begünstigende (prädisponierende) Faktoren. Die ED beginnt in der späten Wachstumsphase bei vier bis acht Monate alten Jungtieren mit einer schmerzhaften Veränderung des Gelenks und der gelenkbildenden Knochenteile (Osteoarthrose) mit Lahmheit.
Der Bewegungsumfang des Ellbogengelenks ist eingeschränkt. Frühzeichen sind Steifigkeit am Morgen oder nach Ruhepausen. Die Krankheit schreitet lebenslang fort und ist nicht heilbar, eine weitgehende Schmerzfreiheit kann aber in vielen Fällen erreicht werden.
Vorkommen und Ursachen
Eine Ellbogendysplasie kann bei allen großwüchsigen Hunderassen auftreten.
Am häufigsten betroffen sind Chow-Chow, Rottweiler, Berner Sennenhund, Grosser Schweizer Sennenhund, Neufundländer, Labrador Retriever, Deutscher Schäferhund und Bordeaux-Dogge. Die Häufigkeit des Auftretens (Prävalenz) beträgt bei einigen Rassen über 40 %.
Die ED wird polygenetisch (über mehrere Gene) vererbt. Der genaue Erbgang und die beteiligten Gene sind bislang nicht bekannt, so dass kein Gentest für die Erkrankung existiert. Der Nachweis kann daher bislang nur über die tierärztliche Beurteilung des Einzeltieres erfolgen, einige Hundezuchtverbände fordern eine Röntgenuntersuchung für Zuchttiere. Der Grad der Vererbbarkeit (Heritabilität) ist für Rüden größer als für Hündinnen und wird je nach Rasse und Population mit Werten zwischen 0,1 und 0,7 angegeben.
Klinische Symptome
Die erkrankten Tiere werden durch Lahmheiten im Bereich der Vordergliedmaße auffällig.
Es besteht hierbei eine Mischform aus Hangbein- und Stützbeinlahmheit, häufig kommt es zu einer Wegführung des Unterarmes und der Pfote von der normalen Achse der Gliedmaße (Abduktion) sowie einem Heranziehen des Ellenbogens an den Körper (Adduktion), wobei die Gliedmaße eingedreht wird. Bei der klinischen Untersuchung kann häufig eine vermehrte Füllung der Gelenkkapsel festgestellt werden, das Gelenk ist meist schmerzhaft und teilweise können Knirschgeräusche wie Pseudokrepitationen ausgelöst werden.
Manifestationen
Eine Ellbogendysplasie entsteht, wenn die gelenkbildenden Knochenteile Oberarmknochen (Humerus), Elle (Ulna) und Speiche (Radius) nicht exakt genug zueinander passen.
Die ungenaue Passform oder Inkongruenz führt zu chronischen Umbauvorgängen am Ellbogengelenk und den gelenkbildenden Knochenteilen (Osteoarthrose), die zu einer Sklerosierung der Knochen und zur Ausbildung von Knochenauswüchsen (Osteophyten) führen. Bei geringer Inkongruenz der Gelenkflächen ist die Osteoarthrose das einzige Anzeichen einer Ellbogendysplasie, darüber hinaus können weitere Veränderungen auftreten:
Fragmentierung des Processus coronoideus medialis (FCP, Ablösung des innen liegenden Kronfortsatzes der Elle).
Osteochondrosis dissecans am Condylus medialis humeri (OCD, Knorpelablösung am innen liegenden Rollhöcker des Oberarmknochens).
Isolierung des Processus anconaeus (IPA, Ablösung des Ellenbogenfortsatzes der Elle).
Ein gleichzeitiges Auftreten mehrerer dieser Komplikationen ist häufig. In der neueren Literatur werden FCP und OCD auch unter dem Begriff Medial Compartment Disease (MCD) zusammengefasst. Gelegentlich werden im deutschsprachigen Raum auch weitere Entwicklungsstörungen wie die ausbleibende Fusion der drei ellenbogenseitigen Verknöcherungskerne des Oberarmknochens und die angeborene Ellbogenluxation oder Subluxation bei kleinen (sogenannten chondrodystrophen) Hunderassen in den Ellbogendysplasie-Komplex eingeordnet.
Letztere begünstigen ebenfalls das Auftreten eines IPA oder FCP, werden aber von der International Elbow working Group nicht zum ED-Komplex gezählt.
Fragmentierung des Processus coronoideus medialis ulnae (FCP)
Als Ursache für die Ablösung des Processus coronoideus medialis (engl. fragmented coronoid process, FCP) werden verschiedene Mechanismen diskutiert:
Wachstumsverzögerung der Speiche mit Verkürzung derselben (short-radius-syndrome), die zu einer verstärkten Belastung der Elle führt. Am Processus coronoideus medialis kommt es dadurch zur Knochenverdichtung (Sklerosierung).
Deformation und schließlich zur Ablösung.
Verfrühter Epiphysenfugenschluss des Radiuskopfes.
Gestörte Feindurchblutung (Mikrovaskularisation) durch eine mechanisch induzierte Sklerose des Knochens im Bereich des Kronfortsatzes.
Die Erkrankung tritt frühestens im Alter von fünf bis sieben Monaten auf. Unter Umständen wird sie aber vom Besitzer nicht sofort bemerkt, so dass auch Tiere erst im zweiten Lebensjahr dem Tierarzt vorgestellt werden. Klinisch äußert sich eine FCP als Lahmheit, die vor allem nach längerer Ruhe oder stärkerer Belastung auftritt. Der Ellenbogen wird zur Seite ausgestellt.
Bei der klinischen Untersuchung zeigt sich eine Schmerzhaftigkeit bei starker Streckung oder Beugung des Gelenks. Im Röntgenbild sind vor allem die Verschattungen im Bereich der Elle, der Verlust der Knochenbälkchenzeichnung, eine undeutliche vordere Kontur im latero-lateralen Strahlengang (seitliche Projektion) und gegebenenfalls die Frakturlinie des Fortsatzes sichtbar. Ein vollständiger Abriss des Processus coronoideus ist jedoch selten.
Aufgrund der mangelnden Kongruenz können sich eine Stufe zwischen Speiche und Elle und ein ungleichmäßig breiter Gelenkspalt darstellen. Diese Inkongruenz lässt sich mittels eines Quotienten darstellen. Hierzu wird die Länge der Incisura trochlearis sowie die Entfernung zwischen der Spitze des Processus anconaeus und der Spitze des Processus coronoideus lateralis ulnae gemessen. Liegt der Quotient beider Werte über 1,15, gilt das Ellenbogengelenk als inkongruent.
Die mit dem FCP verbundene Arthrose zeigt sich bei schwereren Formen in Lippenbildungen der angrenzenden Knochenkonturen. Knochenanbauten treten vor allem am innen liegenden (medialen) Rand der Elle und des Oberarmknochens auf. Eine sichere Diagnose eines FCP ist am Röntgenbild nur selten möglich.
Eine Computertomografie und Arthroskopie kann die Diagnose FCP untermauern.
Osteochondrosis dissecans humeri
Eine Osteochondrosis dissecans (OCD) kommt im Bereich des Ellenbogengelenks fast ausschließlich am innen liegenden Rollhöcker des Oberarmknochens (Condylus medialis humeri) vor. Sie entsteht zumeist im Alter von 5 Monaten und in der Regel beidseitig. Häufiger betroffene Rassen sind Labrador Retriever, Golden Retriever und Rottweiler.
Häufig ist diese Form der Ellbogendysplasie mit einem fragmentierten Processus coronoideus verbunden.
Allerdings wird meist eine „echte“ OCD mit den Knorpelerosionen (kissing lesions) bei einem FCP verwechselt, welche nicht das unter dem Gelenkknorpel gelegene (subchondrale) Knochengewebe betreffen, so dass Read ein gleichzeitiges Auftreten beider Läsionen anzweifelt.
Die Diagnose lässt sich zumeist anhand eines Röntgenbildes, vor allem im anterior-posterioren Strahlengang (Projektion von vorn nach hinten) stellen.
Der röntgenologische Nachweis gelingt jedoch nicht immer, so dass der sichere Ausschluss nur über eine Arthroskopie oder Computertomografie (CT) erfolgen kann.
Isolierter Processus anconaeus (IPA)
Ein selbstständiger (isolierter) Ellenbogenfortsatz der Elle ist eine erblich bedingte Störung der enchondralen Ossifikation und wurde 1956 erstmals beschrieben. Beim IPA unterbleibt die Fusion zwischen Elle und ihrem Processus anconaeus, der ein eigenes Ossifikationszentrum besitzt, welches normalerweise im Alter von 18 bis 24 Wochen mit der Elle verschmilzt.
In diesem Alter besteht aufgrund einer verminderten Elastizität die Gefahr eines teilweisen oder vollständigen Abrisses durch ein Trauma oder das Ausbleiben des Fugenschlusses infolge hoher körperlicher Aktivität. Als weitere Ursache wird ein vermindertes Längenwachstum der Elle (sog. short-ulna-syndrome) diskutiert. Eine Überversorgung mit Calcium und Phosphor begünstigt das Auftreten eines IPA.
Überdurchschnittlich betroffen sind Rottweiler und Deutscher Schäferhund. Bei Rüden ist die Erkrankung häufiger als bei Hündinnen. In etwa 60 % der Fälle tritt ein IPA einseitig auf.
Die Diagnose wird anhand eines Röntgenbildes in Beugestellung des Gelenks gestellt, wobei zu beachten ist, dass der Processus anconeus erst mit etwa sechs Monaten mit der Elle verschmilzt.
Die Frakturlinie ist in den meisten Fällen gut sichtbar, außerdem kommt es zu Sklerosierungen des betroffenen Bereiches und bei längerem Bestehen zu Knochenanbauten.
Therapie
Losgelöste Skelett- (FCP und IPA) bzw. Knorpelteile (OCD) sollten chirurgisch entfernt werden, da sie einen ständigen Reiz auf die Gelenkkapsel ausüben. Diese Entfernung sollte möglichst früh erfolgen, also bevor sich eine Arthrose entwickelt.
Ein IPA kann auch mittels Osteosynthese wieder fixiert werden. Anschließend sollte das Tier zwei bis vier Wochen möglichst gar nicht bewegt werden (Leinenzwang, Boxenruhe) und ihm anschließend für die gleiche Zeit nur wenig Bewegungsspielraum eingeräumt werden. Besteht zusätzlich eine Inkongruenz im Gelenk, ist die chirurgische Entfernung der losgelösten Fragmente allein nicht ausreichend. Hier wird zumeist ein chirurgisches Durchtrennen (Osteotomie) der Elle durchgeführt.
Alle operativen Maßnahmen verhindern jedoch häufig nicht das Fortschreiten der Arthrose. Eine Nutzung als Begleithund ist zwar möglich, von stärkerer Arbeit wie bei Gebrauchshunden wird aber abgeraten.
Bei schweren Ellbogendysplasien kann eine Endoprothese angezeigt sein. Bei sehr starken Veränderungen muss eine Endoprothese oder eine Versteifung des Ellbogengelenks in Erwägung gezogen werden.
Unterstützend ist eine schmerz- und entzündungshemmende Therapie sinnvoll. Hier werden zumeist nichtsteroidale Antiphlogistika wie Carprofen eingesetzt. Eine 2006 veröffentlichte Studie zeigte eine gute Verträglichkeit einer zweimonatigen Therapie mit Carprofen bei Hunden und keine Hinweise auf eine Toxizität für Nieren oder Leber.
Eine Gewichtsreduktion ist bei übergewichtigen Tieren unbedingt zu empfehlen.
Die Wirksamkeit alternativmedizinischer Behandlungsformen (Akupunktur, Gold-Implantate, Homöopathie) ist bislang nicht durch randomisierte Kontrollstudien bewiesen worden. Eine aktuelle evidenzbasierte Studie konnte keine positive Wirkung einer Elektroakupunktur nachweisen.
Eine tägliche Verabreichung von Gelatine als Granulat ins Futter soll eine Arthrose verhindern oder zumindest hinauszögern können.
Vorbeugung und Züchterische Maßnahmen
Hunde mit einer ED sollten wegen der Vererbbarkeit von der Zucht ausgeschlossen werden.
Zur weiteren Bekämpfung in der Zucht wird von einigen Verbänden eine Zuchtwertschätzung durchgeführt. Dazu werden Röntgenaufnahmen von anerkannten Gutachtern beurteilt.
Prinzipiell sind beide Ellbogengelenke zu röntgen und die Tiere müssen zum Zeitpunkt der Untersuchung ein Alter von mindestens 12 Monaten haben.
Zur Befundung sind jeweils eine seitliche Aufnahme (mediolateraler Strahlengang) in 40–90° Beugehaltung sowie eine kraniokaudale in 15° Supination notwendig.
Besser ist es, zwei mediolaterale Aufnahmen mit Beugewinkeln von 30 und von 100–120° zu erstellen, weil sich so ein IPA sicherer nachweisen lässt.
Hohes Körpermassewachstum und Fütterungsfehler sind weitere begünstigende (prädisponierende) Faktoren. Die ED beginnt in der späten Wachstumsphase bei vier bis acht Monate alten Jungtieren mit einer schmerzhaften Veränderung des Gelenks und der gelenkbildenden Knochenteile (Osteoarthrose) mit Lahmheit.
Der Bewegungsumfang des Ellbogengelenks ist eingeschränkt. Frühzeichen sind Steifigkeit am Morgen oder nach Ruhepausen. Die Krankheit schreitet lebenslang fort und ist nicht heilbar, eine weitgehende Schmerzfreiheit kann aber in vielen Fällen erreicht werden.
Vorkommen und Ursachen
Eine Ellbogendysplasie kann bei allen großwüchsigen Hunderassen auftreten.
Am häufigsten betroffen sind Chow-Chow, Rottweiler, Berner Sennenhund, Grosser Schweizer Sennenhund, Neufundländer, Labrador Retriever, Deutscher Schäferhund und Bordeaux-Dogge. Die Häufigkeit des Auftretens (Prävalenz) beträgt bei einigen Rassen über 40 %.
Die ED wird polygenetisch (über mehrere Gene) vererbt. Der genaue Erbgang und die beteiligten Gene sind bislang nicht bekannt, so dass kein Gentest für die Erkrankung existiert. Der Nachweis kann daher bislang nur über die tierärztliche Beurteilung des Einzeltieres erfolgen, einige Hundezuchtverbände fordern eine Röntgenuntersuchung für Zuchttiere. Der Grad der Vererbbarkeit (Heritabilität) ist für Rüden größer als für Hündinnen und wird je nach Rasse und Population mit Werten zwischen 0,1 und 0,7 angegeben.
Klinische Symptome
Die erkrankten Tiere werden durch Lahmheiten im Bereich der Vordergliedmaße auffällig.
Es besteht hierbei eine Mischform aus Hangbein- und Stützbeinlahmheit, häufig kommt es zu einer Wegführung des Unterarmes und der Pfote von der normalen Achse der Gliedmaße (Abduktion) sowie einem Heranziehen des Ellenbogens an den Körper (Adduktion), wobei die Gliedmaße eingedreht wird. Bei der klinischen Untersuchung kann häufig eine vermehrte Füllung der Gelenkkapsel festgestellt werden, das Gelenk ist meist schmerzhaft und teilweise können Knirschgeräusche wie Pseudokrepitationen ausgelöst werden.
Manifestationen
Eine Ellbogendysplasie entsteht, wenn die gelenkbildenden Knochenteile Oberarmknochen (Humerus), Elle (Ulna) und Speiche (Radius) nicht exakt genug zueinander passen.
Die ungenaue Passform oder Inkongruenz führt zu chronischen Umbauvorgängen am Ellbogengelenk und den gelenkbildenden Knochenteilen (Osteoarthrose), die zu einer Sklerosierung der Knochen und zur Ausbildung von Knochenauswüchsen (Osteophyten) führen. Bei geringer Inkongruenz der Gelenkflächen ist die Osteoarthrose das einzige Anzeichen einer Ellbogendysplasie, darüber hinaus können weitere Veränderungen auftreten:
Fragmentierung des Processus coronoideus medialis (FCP, Ablösung des innen liegenden Kronfortsatzes der Elle).
Osteochondrosis dissecans am Condylus medialis humeri (OCD, Knorpelablösung am innen liegenden Rollhöcker des Oberarmknochens).
Isolierung des Processus anconaeus (IPA, Ablösung des Ellenbogenfortsatzes der Elle).
Ein gleichzeitiges Auftreten mehrerer dieser Komplikationen ist häufig. In der neueren Literatur werden FCP und OCD auch unter dem Begriff Medial Compartment Disease (MCD) zusammengefasst. Gelegentlich werden im deutschsprachigen Raum auch weitere Entwicklungsstörungen wie die ausbleibende Fusion der drei ellenbogenseitigen Verknöcherungskerne des Oberarmknochens und die angeborene Ellbogenluxation oder Subluxation bei kleinen (sogenannten chondrodystrophen) Hunderassen in den Ellbogendysplasie-Komplex eingeordnet.
Letztere begünstigen ebenfalls das Auftreten eines IPA oder FCP, werden aber von der International Elbow working Group nicht zum ED-Komplex gezählt.
Fragmentierung des Processus coronoideus medialis ulnae (FCP)
Als Ursache für die Ablösung des Processus coronoideus medialis (engl. fragmented coronoid process, FCP) werden verschiedene Mechanismen diskutiert:
Wachstumsverzögerung der Speiche mit Verkürzung derselben (short-radius-syndrome), die zu einer verstärkten Belastung der Elle führt. Am Processus coronoideus medialis kommt es dadurch zur Knochenverdichtung (Sklerosierung).
Deformation und schließlich zur Ablösung.
Verfrühter Epiphysenfugenschluss des Radiuskopfes.
Gestörte Feindurchblutung (Mikrovaskularisation) durch eine mechanisch induzierte Sklerose des Knochens im Bereich des Kronfortsatzes.
Die Erkrankung tritt frühestens im Alter von fünf bis sieben Monaten auf. Unter Umständen wird sie aber vom Besitzer nicht sofort bemerkt, so dass auch Tiere erst im zweiten Lebensjahr dem Tierarzt vorgestellt werden. Klinisch äußert sich eine FCP als Lahmheit, die vor allem nach längerer Ruhe oder stärkerer Belastung auftritt. Der Ellenbogen wird zur Seite ausgestellt.
Bei der klinischen Untersuchung zeigt sich eine Schmerzhaftigkeit bei starker Streckung oder Beugung des Gelenks. Im Röntgenbild sind vor allem die Verschattungen im Bereich der Elle, der Verlust der Knochenbälkchenzeichnung, eine undeutliche vordere Kontur im latero-lateralen Strahlengang (seitliche Projektion) und gegebenenfalls die Frakturlinie des Fortsatzes sichtbar. Ein vollständiger Abriss des Processus coronoideus ist jedoch selten.
Aufgrund der mangelnden Kongruenz können sich eine Stufe zwischen Speiche und Elle und ein ungleichmäßig breiter Gelenkspalt darstellen. Diese Inkongruenz lässt sich mittels eines Quotienten darstellen. Hierzu wird die Länge der Incisura trochlearis sowie die Entfernung zwischen der Spitze des Processus anconaeus und der Spitze des Processus coronoideus lateralis ulnae gemessen. Liegt der Quotient beider Werte über 1,15, gilt das Ellenbogengelenk als inkongruent.
Die mit dem FCP verbundene Arthrose zeigt sich bei schwereren Formen in Lippenbildungen der angrenzenden Knochenkonturen. Knochenanbauten treten vor allem am innen liegenden (medialen) Rand der Elle und des Oberarmknochens auf. Eine sichere Diagnose eines FCP ist am Röntgenbild nur selten möglich.
Eine Computertomografie und Arthroskopie kann die Diagnose FCP untermauern.
Osteochondrosis dissecans humeri
Eine Osteochondrosis dissecans (OCD) kommt im Bereich des Ellenbogengelenks fast ausschließlich am innen liegenden Rollhöcker des Oberarmknochens (Condylus medialis humeri) vor. Sie entsteht zumeist im Alter von 5 Monaten und in der Regel beidseitig. Häufiger betroffene Rassen sind Labrador Retriever, Golden Retriever und Rottweiler.
Häufig ist diese Form der Ellbogendysplasie mit einem fragmentierten Processus coronoideus verbunden.
Allerdings wird meist eine „echte“ OCD mit den Knorpelerosionen (kissing lesions) bei einem FCP verwechselt, welche nicht das unter dem Gelenkknorpel gelegene (subchondrale) Knochengewebe betreffen, so dass Read ein gleichzeitiges Auftreten beider Läsionen anzweifelt.
Die Diagnose lässt sich zumeist anhand eines Röntgenbildes, vor allem im anterior-posterioren Strahlengang (Projektion von vorn nach hinten) stellen.
Der röntgenologische Nachweis gelingt jedoch nicht immer, so dass der sichere Ausschluss nur über eine Arthroskopie oder Computertomografie (CT) erfolgen kann.
Isolierter Processus anconaeus (IPA)
Ein selbstständiger (isolierter) Ellenbogenfortsatz der Elle ist eine erblich bedingte Störung der enchondralen Ossifikation und wurde 1956 erstmals beschrieben. Beim IPA unterbleibt die Fusion zwischen Elle und ihrem Processus anconaeus, der ein eigenes Ossifikationszentrum besitzt, welches normalerweise im Alter von 18 bis 24 Wochen mit der Elle verschmilzt.
In diesem Alter besteht aufgrund einer verminderten Elastizität die Gefahr eines teilweisen oder vollständigen Abrisses durch ein Trauma oder das Ausbleiben des Fugenschlusses infolge hoher körperlicher Aktivität. Als weitere Ursache wird ein vermindertes Längenwachstum der Elle (sog. short-ulna-syndrome) diskutiert. Eine Überversorgung mit Calcium und Phosphor begünstigt das Auftreten eines IPA.
Überdurchschnittlich betroffen sind Rottweiler und Deutscher Schäferhund. Bei Rüden ist die Erkrankung häufiger als bei Hündinnen. In etwa 60 % der Fälle tritt ein IPA einseitig auf.
Die Diagnose wird anhand eines Röntgenbildes in Beugestellung des Gelenks gestellt, wobei zu beachten ist, dass der Processus anconeus erst mit etwa sechs Monaten mit der Elle verschmilzt.
Die Frakturlinie ist in den meisten Fällen gut sichtbar, außerdem kommt es zu Sklerosierungen des betroffenen Bereiches und bei längerem Bestehen zu Knochenanbauten.
Therapie
Losgelöste Skelett- (FCP und IPA) bzw. Knorpelteile (OCD) sollten chirurgisch entfernt werden, da sie einen ständigen Reiz auf die Gelenkkapsel ausüben. Diese Entfernung sollte möglichst früh erfolgen, also bevor sich eine Arthrose entwickelt.
Ein IPA kann auch mittels Osteosynthese wieder fixiert werden. Anschließend sollte das Tier zwei bis vier Wochen möglichst gar nicht bewegt werden (Leinenzwang, Boxenruhe) und ihm anschließend für die gleiche Zeit nur wenig Bewegungsspielraum eingeräumt werden. Besteht zusätzlich eine Inkongruenz im Gelenk, ist die chirurgische Entfernung der losgelösten Fragmente allein nicht ausreichend. Hier wird zumeist ein chirurgisches Durchtrennen (Osteotomie) der Elle durchgeführt.
Alle operativen Maßnahmen verhindern jedoch häufig nicht das Fortschreiten der Arthrose. Eine Nutzung als Begleithund ist zwar möglich, von stärkerer Arbeit wie bei Gebrauchshunden wird aber abgeraten.
Bei schweren Ellbogendysplasien kann eine Endoprothese angezeigt sein. Bei sehr starken Veränderungen muss eine Endoprothese oder eine Versteifung des Ellbogengelenks in Erwägung gezogen werden.
Unterstützend ist eine schmerz- und entzündungshemmende Therapie sinnvoll. Hier werden zumeist nichtsteroidale Antiphlogistika wie Carprofen eingesetzt. Eine 2006 veröffentlichte Studie zeigte eine gute Verträglichkeit einer zweimonatigen Therapie mit Carprofen bei Hunden und keine Hinweise auf eine Toxizität für Nieren oder Leber.
Eine Gewichtsreduktion ist bei übergewichtigen Tieren unbedingt zu empfehlen.
Die Wirksamkeit alternativmedizinischer Behandlungsformen (Akupunktur, Gold-Implantate, Homöopathie) ist bislang nicht durch randomisierte Kontrollstudien bewiesen worden. Eine aktuelle evidenzbasierte Studie konnte keine positive Wirkung einer Elektroakupunktur nachweisen.
Eine tägliche Verabreichung von Gelatine als Granulat ins Futter soll eine Arthrose verhindern oder zumindest hinauszögern können.
Vorbeugung und Züchterische Maßnahmen
Hunde mit einer ED sollten wegen der Vererbbarkeit von der Zucht ausgeschlossen werden.
Zur weiteren Bekämpfung in der Zucht wird von einigen Verbänden eine Zuchtwertschätzung durchgeführt. Dazu werden Röntgenaufnahmen von anerkannten Gutachtern beurteilt.
Prinzipiell sind beide Ellbogengelenke zu röntgen und die Tiere müssen zum Zeitpunkt der Untersuchung ein Alter von mindestens 12 Monaten haben.
Zur Befundung sind jeweils eine seitliche Aufnahme (mediolateraler Strahlengang) in 40–90° Beugehaltung sowie eine kraniokaudale in 15° Supination notwendig.
Besser ist es, zwei mediolaterale Aufnahmen mit Beugewinkeln von 30 und von 100–120° zu erstellen, weil sich so ein IPA sicherer nachweisen lässt.
eingestellt von: ise; Quellen- und Bildnachweis
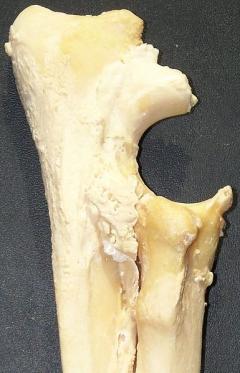
Exercise Induced Collapse

Exercise Induced Collapse (EIC) ist eine erblich bedingte neuromuskuläre Erkrankung, die bei bestimmten Hunderassen beobachtet wurde.
Symptome
Von EIC betroffene Hunde können leicht- bis mittelgradige Anstrengung ohne Probleme tolerieren.
Nach längerer (ab etwa 20 min) starker Anstrengung entwickeln sie jedoch, zumeist ausgehend von der Hinterhand, einen unnatürlichen schwankenden Gang, der sich auf die Vorderhand ausweiten und bis zum Kollaps des Tiers verstärken kann.
Während solcher Episoden erscheinen die meisten Hunde bewusstseinsklar und auch schmerzfrei, gelegentlich wird aber auch ein eingetrübtes Bewusstsein beschrieben. Nach etwa 5- bis 25minütiger Ruhe erholen sich die Tiere in der Regel vollständig.
Vorkommen
EIC tritt hauptsächlich beim Labrador Retriever auf, wurde jedoch auch bei einigen weiteren Hunderassen (bisher: Chesapeake Bay Retriever, Curly Coated Retriever, Boykin Spaniel, Welsh Corgi Pembroke und Deutsch-Drahthaar) beobachtet.
Es scheinen gleichermaßen Hunde aus Arbeits- bzw. Field-Trial-Zuchtlinien als auch solche aus Show-Linien betroffen zu sein. Ein Zusammenhang mit dem Geschlecht oder der Fellfärbung der Tiere zeigt sich nicht.
Die meisten betroffenen Hunde sind bei Erstdiagnose zwischen einem halben und etwa drei Jahren alt.
Genetik
Seit 2007 ist bekannt, dass die für EIC verantwortliche Mutation auf Chromosom 9 lokalisiert ist. Es handelt sich um eine Mutation des Gens für Dynamin-1 (DNM-1).
Diagnostik
Bis zur Verfügbarkeit eines DNA-Tests 2008 konnte EIC nur durch Ausschluss aller anderen Krankheiten mit ähnlicher Symptomatik, etwa Hitzschlag, maligne Hyperthermie, CNM oder bestimmte Formen der Epilepsie festgestellt werden. Die für EIC verantwortliche Mutation des DNM-1-Gens kann per Gentest nachgewiesen werden.
Therapie
Es ist derzeit keine etablierte Therapie bekannt, in einigen Fällen wurde jedoch nach Gabe von Phenobarbital eine Besserung beschrieben.
Von EIC betroffene Tiere sollten generell keinen extremen Belastungen ausgesetzt werden, können jedoch problemlos als Familienhund gehalten werden. Auch artgerechtes Training (Apportieren bzw. Dummytraining, Agility, Jogging) ist in der Regel möglich. In jedem Fall aber muss der Hund beim Auftreten der ersten Symptome einer EIC-Episode ruhig gehalten werden.
Auswirkungen auf die Zucht
EIC wird autosomal-rezessiv vererbt, das heißt Hunde können nur dann erkranken, wenn sie die entsprechende Gen-Mutation von beiden Elterntieren geerbt haben („homozygot betroffen“, „affected“ oder „E/E“).
Bei nur von einem Elternteil geerbter Gen-Mutation („Träger“, „carrier“ bzw. „N/E“) kann der betreffende Hund selbst nicht erkranken, je nach Paarung können unter seinen Nachkommen jedoch ebenfalls Träger oder sogar betroffene Tiere sein. Hunde, die die Mutation von keinem Elternteil geerbt haben („frei“, „clear“ bzw. „N/N“), können weder selbst erkranken noch können unter ihren Nachkommen EIC-betroffene Hunde vorkommen.
Solange sichergestellt ist, dass eines der beiden Elterntiere frei von EIC ist („clear“ bzw. „N/N“), können folglich auch Träger und sogar betroffene Tiere für die Zucht eingesetzt werden.
Weder das Veterinary Diagnostics Laboratory der University of Minnesota (Entwickler des Gentests) noch die Zuchtverbände empfehlen die ausschließliche Verpaarung von N/N-Tieren, denn damit würden auch viele der erwünschten Eigenschaften von N/E- und E/E-Tieren aus der Zucht herausgenommen.
Symptome
Von EIC betroffene Hunde können leicht- bis mittelgradige Anstrengung ohne Probleme tolerieren.
Nach längerer (ab etwa 20 min) starker Anstrengung entwickeln sie jedoch, zumeist ausgehend von der Hinterhand, einen unnatürlichen schwankenden Gang, der sich auf die Vorderhand ausweiten und bis zum Kollaps des Tiers verstärken kann.
Während solcher Episoden erscheinen die meisten Hunde bewusstseinsklar und auch schmerzfrei, gelegentlich wird aber auch ein eingetrübtes Bewusstsein beschrieben. Nach etwa 5- bis 25minütiger Ruhe erholen sich die Tiere in der Regel vollständig.
Vorkommen
EIC tritt hauptsächlich beim Labrador Retriever auf, wurde jedoch auch bei einigen weiteren Hunderassen (bisher: Chesapeake Bay Retriever, Curly Coated Retriever, Boykin Spaniel, Welsh Corgi Pembroke und Deutsch-Drahthaar) beobachtet.
Es scheinen gleichermaßen Hunde aus Arbeits- bzw. Field-Trial-Zuchtlinien als auch solche aus Show-Linien betroffen zu sein. Ein Zusammenhang mit dem Geschlecht oder der Fellfärbung der Tiere zeigt sich nicht.
Die meisten betroffenen Hunde sind bei Erstdiagnose zwischen einem halben und etwa drei Jahren alt.
Genetik
Seit 2007 ist bekannt, dass die für EIC verantwortliche Mutation auf Chromosom 9 lokalisiert ist. Es handelt sich um eine Mutation des Gens für Dynamin-1 (DNM-1).
Diagnostik
Bis zur Verfügbarkeit eines DNA-Tests 2008 konnte EIC nur durch Ausschluss aller anderen Krankheiten mit ähnlicher Symptomatik, etwa Hitzschlag, maligne Hyperthermie, CNM oder bestimmte Formen der Epilepsie festgestellt werden. Die für EIC verantwortliche Mutation des DNM-1-Gens kann per Gentest nachgewiesen werden.
Therapie
Es ist derzeit keine etablierte Therapie bekannt, in einigen Fällen wurde jedoch nach Gabe von Phenobarbital eine Besserung beschrieben.
Von EIC betroffene Tiere sollten generell keinen extremen Belastungen ausgesetzt werden, können jedoch problemlos als Familienhund gehalten werden. Auch artgerechtes Training (Apportieren bzw. Dummytraining, Agility, Jogging) ist in der Regel möglich. In jedem Fall aber muss der Hund beim Auftreten der ersten Symptome einer EIC-Episode ruhig gehalten werden.
Auswirkungen auf die Zucht
EIC wird autosomal-rezessiv vererbt, das heißt Hunde können nur dann erkranken, wenn sie die entsprechende Gen-Mutation von beiden Elterntieren geerbt haben („homozygot betroffen“, „affected“ oder „E/E“).
Bei nur von einem Elternteil geerbter Gen-Mutation („Träger“, „carrier“ bzw. „N/E“) kann der betreffende Hund selbst nicht erkranken, je nach Paarung können unter seinen Nachkommen jedoch ebenfalls Träger oder sogar betroffene Tiere sein. Hunde, die die Mutation von keinem Elternteil geerbt haben („frei“, „clear“ bzw. „N/N“), können weder selbst erkranken noch können unter ihren Nachkommen EIC-betroffene Hunde vorkommen.
Solange sichergestellt ist, dass eines der beiden Elterntiere frei von EIC ist („clear“ bzw. „N/N“), können folglich auch Träger und sogar betroffene Tiere für die Zucht eingesetzt werden.
Weder das Veterinary Diagnostics Laboratory der University of Minnesota (Entwickler des Gentests) noch die Zuchtverbände empfehlen die ausschließliche Verpaarung von N/N-Tieren, denn damit würden auch viele der erwünschten Eigenschaften von N/E- und E/E-Tieren aus der Zucht herausgenommen.
eingestellt von: ise; Quellen- und Bildnachweis

Fibrinoide Leukodystrophie
Die Fibrinoide Leukodystrophie (Alexander's Disease) ist eine sehr selten auftretende Erkrankung bei Labrador Retrievern, Scottish Terriern und Zwergpudeln. Die Ursache ist ungeklärt.
Die Erkrankung führt zu einer Degeneration der Astrozyten. Sie beginnt zwischen drittem und sechstem Lebensmonat mit Hinterhandparese, Ataxie und zunehmender Schwäche. Es können auch Verhaltensänderungen und bei Scottish Terriern Anfälle auftreten.
Die Erkrankung führt zu einer Degeneration der Astrozyten. Sie beginnt zwischen drittem und sechstem Lebensmonat mit Hinterhandparese, Ataxie und zunehmender Schwäche. Es können auch Verhaltensänderungen und bei Scottish Terriern Anfälle auftreten.
eingestellt von: ise; Quellennachweis
Fucosidose
Fucosidose, häufig auch Fukosidose geschrieben oder als α-L-Fucosidase-Mangel bezeichnet, ist eine sehr seltene autosomal-rezessiv vererbte lysosomale Speicherkrankheit aus der Gruppe der Oligosaccharidosen.
Ätiologie
Bei den von der Fucosidose betroffenen Patienten ist das Enzym α-L-Fucosidase in seiner Aktivität vermindert. Ursache für den Enzym-Defekt sind Nonsense- oder Missense-Mutationen auf dem für α-L-Fucosidase kodierenden Gen (FUCA1), welches sich auf Chromosom 1 Genlocus p34 befindet.
Das Enzym katalysiert die Spaltung von L-Fucose, beziehungsweise von Glycolipiden und Oligosacchariden, die Fucose enthalten. Das Substrat Fucose ist ein essentielles Monosaccharid; genauer eine Hexose. Die durch den Gendefekt verursachte Verminderung in der Enzymaktivität führt zu einer Anreicherung der nicht metabolisierten Fucose, beziehungsweise fucosehaltigen Verbindungen, in den Zellen in allen Geweben des Körpers. Diese Ansammlungen führen zu Schädigungen in der Zelle und der betroffenen Organe.
Beim Menschen liegt auf Chromosom 2 ein Pseudogen von FUCA1 und auf Chromosom 6 das polymorphe FUCA2-Gen. Das Genprodukt von FUCA2 kontrolliert die Enzymaktivität im Blutserum und in Fibroblasten. Bisher wurden über 20 unterschiedliche Mutationen von FUCA1 gefunden, die eine Fucosidose auslösen können.
Prävalenz
Der α-L-Fucosidase-Mangel ist eine sehr seltene Erkrankung, dessen Prävalenz etwa bei 1 : 1 Million liegt. Weltweit wurden bisher weniger als 100 Patienten beschrieben, wovon etwa 20 aus Süditalien in den Ortschaften Grotteria und Mammola im Bereich von Reggio Calabria stammen.
Eine weitere Patientenhäufung findet sich im Südwesten der Vereinigten Staaten (New Mexico und Colorado). Bei den bisher bekannten 45 betroffenen Familien besteht zu 40 % Blutsverwandtschaft. Die hohe Prävalenz in diesen Populationen lässt sich teilweise durch den Gründereffekt erklären.
Symptomatik und Diagnostik
Es wird zwischen zwei unterschiedlichen Verlaufsformen der Erkrankung unterschieden, wobei die Übergänge zwischen beiden Formen fließend sind. Der klinisch schwerere Typ 1 beginnt im Alter zwischen 3 und 18 Monaten.
Der Typ 2 beginnt zwischen dem 12 und 24 Monat und ist durch einen weniger progressiven Krankheitsverlauf gekennzeichnet.
Die Lebenserwartung beim Typ 2 ist höher als bei Patienten mit Typ 1. Daneben wird noch ein dritter Typus der Erkrankung diskutiert, der bei zwei niederländischen Patienten beschrieben wurde. Es handelt sich um eine weniger stark ausgeprägte, mehr juvenile Form, der Erkrankung, bei der keine Angiokeratome auftreten.
Typische Symptome der Fucosidose sind Deformationen des Gesichts (Gesichtsdysmorphie) und Fehlbildungen des Skeletts, schwere geistige Retardierung, Krampfanfälle, Vergrößerung der Leber (Hepatomegalie), Milz (Splenomegalie) und des Herzens (Kardiomegalie). Daneben sind noch Gehörlosigkeit und Angiokeratome zu beobachten.
Diagnostisch lässt sich die Fucosidose durch chromatografische Analyse des Urins der Patienten bestimmen. Die Aktivitätsbestimmung der α-L-Fucosidase in den Leukozyten kann im Labor den Befund absichern. Ein Gentest ist in den meisten Fällen nicht notwendig.
Therapie
Die bisher einzige, nicht-symptomatische, Behandlung mit kurativem Ansatz ist die Knochenmarkstransplantation. Da bisher weltweit erst zehn Patienten so behandelt wurden, kann dieser Therapieansatz noch nicht abschließend beurteilt werden.
Die Methode wurde zuerst an Hunden erfolgreich erprobt.
Ätiologie
Bei den von der Fucosidose betroffenen Patienten ist das Enzym α-L-Fucosidase in seiner Aktivität vermindert. Ursache für den Enzym-Defekt sind Nonsense- oder Missense-Mutationen auf dem für α-L-Fucosidase kodierenden Gen (FUCA1), welches sich auf Chromosom 1 Genlocus p34 befindet.
Das Enzym katalysiert die Spaltung von L-Fucose, beziehungsweise von Glycolipiden und Oligosacchariden, die Fucose enthalten. Das Substrat Fucose ist ein essentielles Monosaccharid; genauer eine Hexose. Die durch den Gendefekt verursachte Verminderung in der Enzymaktivität führt zu einer Anreicherung der nicht metabolisierten Fucose, beziehungsweise fucosehaltigen Verbindungen, in den Zellen in allen Geweben des Körpers. Diese Ansammlungen führen zu Schädigungen in der Zelle und der betroffenen Organe.
Beim Menschen liegt auf Chromosom 2 ein Pseudogen von FUCA1 und auf Chromosom 6 das polymorphe FUCA2-Gen. Das Genprodukt von FUCA2 kontrolliert die Enzymaktivität im Blutserum und in Fibroblasten. Bisher wurden über 20 unterschiedliche Mutationen von FUCA1 gefunden, die eine Fucosidose auslösen können.
Prävalenz
Der α-L-Fucosidase-Mangel ist eine sehr seltene Erkrankung, dessen Prävalenz etwa bei 1 : 1 Million liegt. Weltweit wurden bisher weniger als 100 Patienten beschrieben, wovon etwa 20 aus Süditalien in den Ortschaften Grotteria und Mammola im Bereich von Reggio Calabria stammen.
Eine weitere Patientenhäufung findet sich im Südwesten der Vereinigten Staaten (New Mexico und Colorado). Bei den bisher bekannten 45 betroffenen Familien besteht zu 40 % Blutsverwandtschaft. Die hohe Prävalenz in diesen Populationen lässt sich teilweise durch den Gründereffekt erklären.
Symptomatik und Diagnostik
Es wird zwischen zwei unterschiedlichen Verlaufsformen der Erkrankung unterschieden, wobei die Übergänge zwischen beiden Formen fließend sind. Der klinisch schwerere Typ 1 beginnt im Alter zwischen 3 und 18 Monaten.
Der Typ 2 beginnt zwischen dem 12 und 24 Monat und ist durch einen weniger progressiven Krankheitsverlauf gekennzeichnet.
Die Lebenserwartung beim Typ 2 ist höher als bei Patienten mit Typ 1. Daneben wird noch ein dritter Typus der Erkrankung diskutiert, der bei zwei niederländischen Patienten beschrieben wurde. Es handelt sich um eine weniger stark ausgeprägte, mehr juvenile Form, der Erkrankung, bei der keine Angiokeratome auftreten.
Typische Symptome der Fucosidose sind Deformationen des Gesichts (Gesichtsdysmorphie) und Fehlbildungen des Skeletts, schwere geistige Retardierung, Krampfanfälle, Vergrößerung der Leber (Hepatomegalie), Milz (Splenomegalie) und des Herzens (Kardiomegalie). Daneben sind noch Gehörlosigkeit und Angiokeratome zu beobachten.
Diagnostisch lässt sich die Fucosidose durch chromatografische Analyse des Urins der Patienten bestimmen. Die Aktivitätsbestimmung der α-L-Fucosidase in den Leukozyten kann im Labor den Befund absichern. Ein Gentest ist in den meisten Fällen nicht notwendig.
Therapie
Die bisher einzige, nicht-symptomatische, Behandlung mit kurativem Ansatz ist die Knochenmarkstransplantation. Da bisher weltweit erst zehn Patienten so behandelt wurden, kann dieser Therapieansatz noch nicht abschließend beurteilt werden.
Die Methode wurde zuerst an Hunden erfolgreich erprobt.
eingestellt von: ise; Quellennachweis
Hüftdysplasie des Hundes

Die Hüftdysplasie oder Hüftgelenksdysplasie (HD) ist eine Fehlentwicklung des Hüftgelenks. Betroffen sind sämtliche Hunderassen, wobei großwüchsige Rassen das Krankheitsbild besonders häufig ausbilden.
Erstmals diagnostiziert wurde sie am Deutschen Schäferhund und wird daher fälschlicherweise hauptsächlich mit dieser Rasse in Verbindung gebracht, obwohl mittlerweile andere Rassen stärker betroffen sind. Die Häufigkeit des Vorkommens (Prävalenz) kann je nach Rasse bis über 50 Prozent betragen. Auch bei Hauskatzen kann diese Krankheit auftreten, besonders unter Maine-Coon-Katzen.
Die HD ist zu großen Teilen genetisch bedingt (die Heritabilität liegt zwischen 20 und 40 Prozent), weshalb viele Zuchtverbände die HD-Freiheit zur Zuchtzulassung fordern.
Da falsche Ernährung und Haltung die Entstehung und das Fortschreiten der Krankheit begünstigen können, handelt es sich um ein multifaktorielles (von vielen Faktoren abhängiges) Geschehen. Klinisch zeigt sich die HD in zunehmender Bewegungseinschränkung und Schmerzhaftigkeit, die infolge der krankhaften Umbauprozesse am Hüftgelenk (Coxarthrose) entstehen. Im fortgeschrittenen Stadium kann nur die Entfernung des Hüftgelenks mit oder ohne Einsetzen eines künstlichen Hüftgelenks eine deutliche Verbesserung herbeiführen. Ist dies nicht möglich, lässt sich durch eine dauerhafte Schmerztherapie häufig lange eine ausreichende Lebensqualität aufrechterhalten.
Symptome und Diagnostik
Die Ausprägung klinischer Symptome einer HD variiert in Abhängigkeit vom Alter bzw. Stadium der Krankheit. Bei relativ jungen Tieren, im Alter von einem halben bis einem Jahr, kommt es zu Schmerzen, weil der Kopf des Oberschenkelknochens in der Hüftgelenkspfanne (Acetabulum) nur ungenügenden Halt findet und durch seine abnorme Beweglichkeit schmerzregistrierende Nervenfasern der Knochenhaut des Pfannenrandes gereizt werden. Ältere Tiere bilden Schmerzen eher infolge fortschreitender degenerativer Veränderungen (Arthrosen) des Hüftgelenkes aus.
Eine beginnende HD äußert sich in zunehmenden Schmerzen bei Spaziergängen, der Hund will nicht mehr weit laufen, setzt sich öfter hin, schreit beim Spielen gelegentlich auf und zeigt einen instabilen Gang. Beim Vorführen der Hintergliedmaße wird das Becken in Richtung der vorgeführten Gliedmaße seitwärts bewegt (LSÜ-twist). Bei Bewegungen des Gelenkes kann ein Knacken, Klicken oder Knirschen des Gelenks hörbar sein. Bei Feststellung eines der Symptome ist der sofortige Gang zum Tierarzt ratsam.
Palpation
Bereits über Belastung einzelner Gelenke können auch unklare Lahmheiten der Hintergliedmaße beim Vorliegen einer HD oft rasch dem Hüftgelenk zugeordnet werden. Die Bewertung einer schweren Hüftdysplasie macht häufig spezielle Tests erforderlich, um eine Aussage über die Gelenkstabilität treffen zu können. Am häufigsten wird der Ortolani-Test verwendet:
Der Oberschenkel wird beim auf der gesunden Seite liegenden Tier im rechten Winkel zur Wirbelsäule gelagert. Eine auf das Kniegelenk aufgelegte Hand schiebt nun unter starkem Druck den Oberschenkelknochen in Richtung Wirbelsäule. Bei starker Instabilität des Gelenkes kommt es dadurch zur Luxation oder Subluxation des Hüftgelenkes. Wird nun der Oberschenkel von der Körperachse weggeführt, gleitet der Oberschenkelkopf mit einem Klickgeräusch (Ortolani-Klick) in die Pfanne zurück.
Dieser Test sollte möglichst nur von einem Tierarzt durchgeführt werden, unter Umständen in Narkose.
Röntgen
Eine zuverlässige Möglichkeit, den Schweregrad der Erkrankung zu erkennen, ist die Röntgenuntersuchung. Dabei müssen die Gelenke überstreckt werden, was beim Vorliegen einer HD starke Schmerzen verursacht. Daher wird sie unter Kurznarkose durchgeführt. Voraussetzung für eine aussagekräftige Diagnose ist die exakte Positionierung des Tieres in Rückenlage mit gestreckten, parallel gelagerten Oberschenkeln und rechtwinklig zum Strahlengang eingedrehten Kniescheiben. Zusätzliche Aufnahmen können in „Froschhaltung“ der Oberschenkel oder im seitlichen (latero-lateralen) Strahlengang erfolgen.
Ein wesentliches Auswertungskriterium ist der Norberg-Winkel. Er ist als der Winkel definiert, der zwischen der Verbindungslinie der Zentren der beiden Oberschenkelköpfe und dem vorderen Pfannenrand abgetragen wird (siehe Abbildung). Bei einem HD-freien Tier sollte er mehr als 105° betragen (gelbe Linien). Weitere Beurteilungskriterien sind die Kongruenz von Oberschenkelkopf und Gelenkpfanne, die Weite des Gelenkspaltes, die Pfannenkontur, die Kontur des Oberschenkelkopfes sowie das Vorhandensein von Hinweisen auf arthrotische Prozesse wie walzenförmige Verdickungen des Oberschenkelhalses, Randwülste an der Gelenkpfanne, unter dem Knorpel befindliche Verdichtungen der Knochensubstanz im Pfannenbereich und die Anlagerung von Knochenmaterial (Osteophyt) am Ansatz der Gelenkkapsel (Morgan-Linie). Die Morgan-Linie ist ein sensitiver Frühmarker für eine Instabilität im Hüftgelenk.
Die züchterische Auswertung von HD-Aufnahmen ist im VDH nur durch von den Rassezuchtverbänden zugelassene Gutachter möglich, an die der Tierarzt die Röntgenbilder einschickt.
Differentialdiagnosen
Diagnostisch muss eine Hüftgelenksdysplasie von anderen Störungen des Skelettsystems abgegrenzt werden. Neben Knochenbrüchen und Luxationen sind dies bei großen Hunderassen vor allem Tumore der Knochen, welche im Bereich des Oberschenkelknochens relativ häufig auftreten. Bei kleinwüchsigen Tieren muss die aseptische Femurkopfnekrose (Legg-Calvé-Perthes-Krankheit) abgegrenzt werden.
Weiterhin treten bei schnellwachsenden Hunden häufig Ablösungen des Gelenkknorpels auf (Osteochondrosis dissecans), die ebenfalls schmerzhaft sind. Ferner sind Erkrankungen des Kniegelenks (z. B. Kreuzbandriss), Beckenbrüche und Erkrankungen der Wirbelsäule (Bandscheibenvorfall, vor allem bei kleinen Hunderassen) sowie Instabilität am Lenden-Kreuzbeinübergang der Wirbelsäule (Cauda-equina-Syndrom, häufiger beim Deutschen Schäferhund) auszuschließen.
Behandlung
Man kann HD nicht heilen, sondern nur das Auftreten klinischer Symptome und das Fortschreiten der Krankheit hinauszögern oder die Schmerzen reduzieren. Je häufiger der Hund bestimmte Bewegungsabläufe ausführt, desto schneller verschleißt die Hüfte. Zu diesen Bewegungen gehören vor allem jene, die die Gelenke besonders stauchen, wie Treppenlaufen, Springen auf harten Untergründen und ähnliche. Man kann dem Hund mit frühzeitigem Erkennen und richtigem Umgang mit der Krankheit ein normales Leben ermöglichen.
Es gibt folgende Behandlungsmöglichkeiten:
Medikamentöse Therapie mit entzündungshemmenden und schmerzstillenden Medikamenten (Antiphlogistika)
PIN-Operation: Durchtrennung oder Entfernung des Musculus pectineus sowie Umschneiden des Gelenkkapselrandes zur Unterbindung der schmerzleitenden Nervenfasern. Dies ist eine sehr effektive Schmerztherapie, deren Wirkung mehrere Jahre anhält.
Kapselraffung: Hierbei wird die Gelenkkapsel chirurgisch gestrafft. Die Operation ist nur bei jungen Tieren sinnvoll, wenn noch keine deutlichen Abnutzungserscheinungen aufgetreten sind und verhindert die Subluxationen und damit ein Fortschreiten der Erkrankung.
Osteotomie des Beckens: Dazu werden alle drei Beckenknochen (Darmbein, Sitzbein und Schambein) durchtrennt, das Becken etwas zur Seite gekippt und die Knochen anschließend wieder durch Osteosynthese verbunden. Ziel ist es, dass der Oberschenkelkopf wieder besser zur Hüftgelenkspfanne steht. Diese Operation ist aufwändig und nur bei jungen Hunden anzuraten, bei denen noch keine sichtbaren Veränderungen an der Gestalt des Femurkopfs im Sinne einer beginnenden Arthrose bestehen.
Das Einsetzen eines künstlichen Hüftgelenkes ist eine sehr kostenintensive Behandlung. Die Operation führt im Regelfall zur Beschwerdefreiheit bis ins hohe Alter. Es ist wichtig, den Hund beim anschließenden Muskelaufbau durch viel Bewegung zu unterstützen. Laufen am Fahrrad und Schwimmen sind ideal. Gute Resultate werden auch mit der zusätzlichen Medikation von Muskelaufbaupräparaten erzielt.
Femurkopfresektion: Dabei wird der Gelenkkopf des Oberschenkelknochens (Caput ossis femoris) entfernt, worauf sich eine bindegewebige Verbindung zwischen Becken und Oberschenkelknochen entwickelt. Verbunden mit intensiver Physiotherapie bietet diese Methode gute Chancen, ein schmerzfreies Leben zu führen. Häufig bleibt durch diese Behandlungsmethode jedoch eine dauerhafte Funktionsstörung zurück.
Stammzellentherapie: Mit der Stammzellentherapie ist es möglich, Knorpel wieder aufzubauen und eine Schmerzreduktion herbeizuführen.
Einsetzen von einem oder mehreren Goldstiften in die Muskulatur an Akupunkturpunkten. Die Goldstifte verbleiben in der Muskulatur. Diese Behandlungsmethode ist in den Bereich der Alternativmedizin einzuordnen, ihre Wirksamkeit ist nicht belegt.
Physiotherapie: zur Schmerzlinderung und zum Muskelaufbau.
Orthopädisches Hundebett: zur Schmerzlinderung, eine Wirkung ist jedoch nicht nachgewiesen.
Vorbeugung
Eine Verhinderung des Fortschreitens kann durch richtige Ernährung und nicht zu viel Sport – vor allem durch wenig Belastung und das Vermeiden von Stauchen und Überdehnen des Hüftgelenkes – erreicht werden.
Eine Physiotherapie kann durch den gezielten Aufbau der Becken- und Oberschenkelmuskulatur das Hüftgelenk entlasten. Die Zugabe von knorpelaufbauenden Zusatzfuttermitteln ist ebenfalls möglich.
Zur Vermeidung der Weitervererbung der Fehlbildung ist bei den meisten Hundezuchtverbänden eine Bescheinigung der HD-Freiheit zur Zuchtzulassung erforderlich. Aber auch die Paarung aus HD-freien Elterntieren bietet keine Garantie, dass die Nachkommen HD-frei sind.
Viele Zuchtverbände wenden bei der Selektion auch eine Zuchtwertschätzung an.
Erstmals diagnostiziert wurde sie am Deutschen Schäferhund und wird daher fälschlicherweise hauptsächlich mit dieser Rasse in Verbindung gebracht, obwohl mittlerweile andere Rassen stärker betroffen sind. Die Häufigkeit des Vorkommens (Prävalenz) kann je nach Rasse bis über 50 Prozent betragen. Auch bei Hauskatzen kann diese Krankheit auftreten, besonders unter Maine-Coon-Katzen.
Die HD ist zu großen Teilen genetisch bedingt (die Heritabilität liegt zwischen 20 und 40 Prozent), weshalb viele Zuchtverbände die HD-Freiheit zur Zuchtzulassung fordern.
Da falsche Ernährung und Haltung die Entstehung und das Fortschreiten der Krankheit begünstigen können, handelt es sich um ein multifaktorielles (von vielen Faktoren abhängiges) Geschehen. Klinisch zeigt sich die HD in zunehmender Bewegungseinschränkung und Schmerzhaftigkeit, die infolge der krankhaften Umbauprozesse am Hüftgelenk (Coxarthrose) entstehen. Im fortgeschrittenen Stadium kann nur die Entfernung des Hüftgelenks mit oder ohne Einsetzen eines künstlichen Hüftgelenks eine deutliche Verbesserung herbeiführen. Ist dies nicht möglich, lässt sich durch eine dauerhafte Schmerztherapie häufig lange eine ausreichende Lebensqualität aufrechterhalten.
Symptome und Diagnostik
Die Ausprägung klinischer Symptome einer HD variiert in Abhängigkeit vom Alter bzw. Stadium der Krankheit. Bei relativ jungen Tieren, im Alter von einem halben bis einem Jahr, kommt es zu Schmerzen, weil der Kopf des Oberschenkelknochens in der Hüftgelenkspfanne (Acetabulum) nur ungenügenden Halt findet und durch seine abnorme Beweglichkeit schmerzregistrierende Nervenfasern der Knochenhaut des Pfannenrandes gereizt werden. Ältere Tiere bilden Schmerzen eher infolge fortschreitender degenerativer Veränderungen (Arthrosen) des Hüftgelenkes aus.
Eine beginnende HD äußert sich in zunehmenden Schmerzen bei Spaziergängen, der Hund will nicht mehr weit laufen, setzt sich öfter hin, schreit beim Spielen gelegentlich auf und zeigt einen instabilen Gang. Beim Vorführen der Hintergliedmaße wird das Becken in Richtung der vorgeführten Gliedmaße seitwärts bewegt (LSÜ-twist). Bei Bewegungen des Gelenkes kann ein Knacken, Klicken oder Knirschen des Gelenks hörbar sein. Bei Feststellung eines der Symptome ist der sofortige Gang zum Tierarzt ratsam.
Palpation
Bereits über Belastung einzelner Gelenke können auch unklare Lahmheiten der Hintergliedmaße beim Vorliegen einer HD oft rasch dem Hüftgelenk zugeordnet werden. Die Bewertung einer schweren Hüftdysplasie macht häufig spezielle Tests erforderlich, um eine Aussage über die Gelenkstabilität treffen zu können. Am häufigsten wird der Ortolani-Test verwendet:
Der Oberschenkel wird beim auf der gesunden Seite liegenden Tier im rechten Winkel zur Wirbelsäule gelagert. Eine auf das Kniegelenk aufgelegte Hand schiebt nun unter starkem Druck den Oberschenkelknochen in Richtung Wirbelsäule. Bei starker Instabilität des Gelenkes kommt es dadurch zur Luxation oder Subluxation des Hüftgelenkes. Wird nun der Oberschenkel von der Körperachse weggeführt, gleitet der Oberschenkelkopf mit einem Klickgeräusch (Ortolani-Klick) in die Pfanne zurück.
Dieser Test sollte möglichst nur von einem Tierarzt durchgeführt werden, unter Umständen in Narkose.
Röntgen
Eine zuverlässige Möglichkeit, den Schweregrad der Erkrankung zu erkennen, ist die Röntgenuntersuchung. Dabei müssen die Gelenke überstreckt werden, was beim Vorliegen einer HD starke Schmerzen verursacht. Daher wird sie unter Kurznarkose durchgeführt. Voraussetzung für eine aussagekräftige Diagnose ist die exakte Positionierung des Tieres in Rückenlage mit gestreckten, parallel gelagerten Oberschenkeln und rechtwinklig zum Strahlengang eingedrehten Kniescheiben. Zusätzliche Aufnahmen können in „Froschhaltung“ der Oberschenkel oder im seitlichen (latero-lateralen) Strahlengang erfolgen.
Ein wesentliches Auswertungskriterium ist der Norberg-Winkel. Er ist als der Winkel definiert, der zwischen der Verbindungslinie der Zentren der beiden Oberschenkelköpfe und dem vorderen Pfannenrand abgetragen wird (siehe Abbildung). Bei einem HD-freien Tier sollte er mehr als 105° betragen (gelbe Linien). Weitere Beurteilungskriterien sind die Kongruenz von Oberschenkelkopf und Gelenkpfanne, die Weite des Gelenkspaltes, die Pfannenkontur, die Kontur des Oberschenkelkopfes sowie das Vorhandensein von Hinweisen auf arthrotische Prozesse wie walzenförmige Verdickungen des Oberschenkelhalses, Randwülste an der Gelenkpfanne, unter dem Knorpel befindliche Verdichtungen der Knochensubstanz im Pfannenbereich und die Anlagerung von Knochenmaterial (Osteophyt) am Ansatz der Gelenkkapsel (Morgan-Linie). Die Morgan-Linie ist ein sensitiver Frühmarker für eine Instabilität im Hüftgelenk.
Die züchterische Auswertung von HD-Aufnahmen ist im VDH nur durch von den Rassezuchtverbänden zugelassene Gutachter möglich, an die der Tierarzt die Röntgenbilder einschickt.
Differentialdiagnosen
Diagnostisch muss eine Hüftgelenksdysplasie von anderen Störungen des Skelettsystems abgegrenzt werden. Neben Knochenbrüchen und Luxationen sind dies bei großen Hunderassen vor allem Tumore der Knochen, welche im Bereich des Oberschenkelknochens relativ häufig auftreten. Bei kleinwüchsigen Tieren muss die aseptische Femurkopfnekrose (Legg-Calvé-Perthes-Krankheit) abgegrenzt werden.
Weiterhin treten bei schnellwachsenden Hunden häufig Ablösungen des Gelenkknorpels auf (Osteochondrosis dissecans), die ebenfalls schmerzhaft sind. Ferner sind Erkrankungen des Kniegelenks (z. B. Kreuzbandriss), Beckenbrüche und Erkrankungen der Wirbelsäule (Bandscheibenvorfall, vor allem bei kleinen Hunderassen) sowie Instabilität am Lenden-Kreuzbeinübergang der Wirbelsäule (Cauda-equina-Syndrom, häufiger beim Deutschen Schäferhund) auszuschließen.
Behandlung
Man kann HD nicht heilen, sondern nur das Auftreten klinischer Symptome und das Fortschreiten der Krankheit hinauszögern oder die Schmerzen reduzieren. Je häufiger der Hund bestimmte Bewegungsabläufe ausführt, desto schneller verschleißt die Hüfte. Zu diesen Bewegungen gehören vor allem jene, die die Gelenke besonders stauchen, wie Treppenlaufen, Springen auf harten Untergründen und ähnliche. Man kann dem Hund mit frühzeitigem Erkennen und richtigem Umgang mit der Krankheit ein normales Leben ermöglichen.
Es gibt folgende Behandlungsmöglichkeiten:
Medikamentöse Therapie mit entzündungshemmenden und schmerzstillenden Medikamenten (Antiphlogistika)
PIN-Operation: Durchtrennung oder Entfernung des Musculus pectineus sowie Umschneiden des Gelenkkapselrandes zur Unterbindung der schmerzleitenden Nervenfasern. Dies ist eine sehr effektive Schmerztherapie, deren Wirkung mehrere Jahre anhält.
Kapselraffung: Hierbei wird die Gelenkkapsel chirurgisch gestrafft. Die Operation ist nur bei jungen Tieren sinnvoll, wenn noch keine deutlichen Abnutzungserscheinungen aufgetreten sind und verhindert die Subluxationen und damit ein Fortschreiten der Erkrankung.
Osteotomie des Beckens: Dazu werden alle drei Beckenknochen (Darmbein, Sitzbein und Schambein) durchtrennt, das Becken etwas zur Seite gekippt und die Knochen anschließend wieder durch Osteosynthese verbunden. Ziel ist es, dass der Oberschenkelkopf wieder besser zur Hüftgelenkspfanne steht. Diese Operation ist aufwändig und nur bei jungen Hunden anzuraten, bei denen noch keine sichtbaren Veränderungen an der Gestalt des Femurkopfs im Sinne einer beginnenden Arthrose bestehen.
Das Einsetzen eines künstlichen Hüftgelenkes ist eine sehr kostenintensive Behandlung. Die Operation führt im Regelfall zur Beschwerdefreiheit bis ins hohe Alter. Es ist wichtig, den Hund beim anschließenden Muskelaufbau durch viel Bewegung zu unterstützen. Laufen am Fahrrad und Schwimmen sind ideal. Gute Resultate werden auch mit der zusätzlichen Medikation von Muskelaufbaupräparaten erzielt.
Femurkopfresektion: Dabei wird der Gelenkkopf des Oberschenkelknochens (Caput ossis femoris) entfernt, worauf sich eine bindegewebige Verbindung zwischen Becken und Oberschenkelknochen entwickelt. Verbunden mit intensiver Physiotherapie bietet diese Methode gute Chancen, ein schmerzfreies Leben zu führen. Häufig bleibt durch diese Behandlungsmethode jedoch eine dauerhafte Funktionsstörung zurück.
Stammzellentherapie: Mit der Stammzellentherapie ist es möglich, Knorpel wieder aufzubauen und eine Schmerzreduktion herbeizuführen.
Einsetzen von einem oder mehreren Goldstiften in die Muskulatur an Akupunkturpunkten. Die Goldstifte verbleiben in der Muskulatur. Diese Behandlungsmethode ist in den Bereich der Alternativmedizin einzuordnen, ihre Wirksamkeit ist nicht belegt.
Physiotherapie: zur Schmerzlinderung und zum Muskelaufbau.
Orthopädisches Hundebett: zur Schmerzlinderung, eine Wirkung ist jedoch nicht nachgewiesen.
Vorbeugung
Eine Verhinderung des Fortschreitens kann durch richtige Ernährung und nicht zu viel Sport – vor allem durch wenig Belastung und das Vermeiden von Stauchen und Überdehnen des Hüftgelenkes – erreicht werden.
Eine Physiotherapie kann durch den gezielten Aufbau der Becken- und Oberschenkelmuskulatur das Hüftgelenk entlasten. Die Zugabe von knorpelaufbauenden Zusatzfuttermitteln ist ebenfalls möglich.
Zur Vermeidung der Weitervererbung der Fehlbildung ist bei den meisten Hundezuchtverbänden eine Bescheinigung der HD-Freiheit zur Zuchtzulassung erforderlich. Aber auch die Paarung aus HD-freien Elterntieren bietet keine Garantie, dass die Nachkommen HD-frei sind.
Viele Zuchtverbände wenden bei der Selektion auch eine Zuchtwertschätzung an.
eingestellt von: ise; Quellen- und Bildnachweis
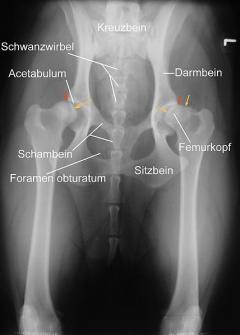
Harnleiterektopie
Die Harnleiterektopie oder Ureterektopie ist eine angeborene Fehlbildung der Harnleiter, die vor allem beim Hund vorkommt und dort bei einigen Rassen erblich zu sein scheint. Hündinnen sind wesentlich häufiger betroffen als Rüden.
Pathophysiologie
Beim gesunden Hund münden die Harnleiter innerhalb des Trigonum vesicae (Harnblasendreieck) in die Harnblase, wo der Urin bis zur Miktion durch den Musculus urethralis zurückbehalten wird.
Bei Harnleiterektopie liegt die Mündung eines oder beider Harnleiter unterhalb dieses Schliessmuskels und mündet statt in die Harnblase in die Harnröhre, die Vagina oder den Uterus. Weil der Urin nicht zurückbehalten werden kann, kommt es zu Harninkontinenz, die sich in ständigem leichtem Harntröpfeln äußert.
Als Komplikationen können lokale Hautreizungen und aufsteigende Harnwegsinfektionen auftreten.
Klinik
Die Krankheit tritt bei Norwegischen Elchhunden, Siberian Huskies, Neufundländern, Golden und Labrador Retrievern, West Highland White Terriers, Foxterriers und Zwergpudeln gehäuft auf, kann aber bei allen Rassen sporadisch auftreten. Hündinnen sind acht mal so oft betroffen wie Rüden.
Die meisten Fälle werden zwischen dem Alter von drei und sechs Monaten vorgestellt.
Symptome
Das Leitsymptom der Harnleiterektopie ist Harninkontinenz mit ständigem Harntröpfeln.
Tiere mit einem normalen und einem ektopischen Harnleiter zeigen zusätzlich auch normales Harnabsatzverhalten, während Tiere mit beidseitig ektopischen Harnleitern nicht normal urinieren können. Durch das ständige Harntröpfeln kann es bei Hündinnen zu Hautreizungen und Infektionen im Bereich der Vagina und Vulva kommen, die bis in die Nieren aufsteigen können.
Rüden entwickeln seltener Symptome, weil sie die Umgehung des M. urethralis normalerweise durch den Musculus sphincter urethrae internus kompensieren können, der die gesamte Länge der Harnröhre umfasst.
Diagnose
Die Diagnose erfolgt über die Injektion eines Kontrastmittels in den Blutkreislauf, das über die Nieren ausgeschieden wird. Mittels Röntgenaufnahmen kann der Weg des Kontrastmittels durch Niere und Harnleiter bis in die Harnblase nachvollzogen werden.
Ist ein ektopischer Harnleiter vorhanden, zeigen die Röntgenbilder dessen Verlauf an der Blase vorbei bis zu seiner Mündung. Gleichzeitig erlaubt diese Diagnosemethode auch die Darstellung von eventuell vorhandenen zusätzlichen Anomalien im Harntrakt, wie etwa einer Hydronephrose.
Therapie und Prognose
Eine kurative Behandlung erfolgt chirurgisch.
Bei einer einseitigen Harnleiterektopie kann eine Nephrektomie der betroffenen Niere durchgeführt werden, insbesondere auch, wenn Komplikationen wie etwa eine Hydronephrose oder Pyelonephritis vorhanden sind. Bei einer beidseitigen Harnleiterektopie ist die Behandlung der Wahl eine Transplantation der ektopischen Harnleitermündungen in die Harnblase, wodurch in den Harnwegen eine funktionell normale Situation erreicht werden kann. Die Transplantation ist auch bei einseitiger Ektopie möglich, falls die Niere der betroffenen Seite normal ist.
Bei einer beidseitigen Harnleiterektopie sowie bei einer einseitigen Ektopie mit einer nicht vollkommen gesunden kontralateralen Niere ist die Nephrektomie kontraindiziert. In solchen Fällen besteht die Behandlung aus der Transplantation der ektopischen Uretermündung in die Harnblase.
In leichten Fällen kann die Harninkontinenz auch symptomatisch behandelt werden, beispielsweise durch Gaben von Phenylpropanolamin oder Ephedrin.
Bei einer erfolgreichen chirurgischen Behandlung ist die Prognose gut. Gelegentlich kommt es zu Komplikationen wie persistierender Harninkontinenz oder Dysurie sowie zu Hydronephrose.
Genetik und Zuchthygiene
Der Erbgang der Harnleiterektopie beim Hund ist nicht bekannt, vermutlich handelt es sich um eine polygene Krankheit. Betroffene Hunde sollten nicht zur Zucht verwendet werden; Verpaarungen, aus denen Welpen mit Harnleiterektopie hervorgingen, sollten nicht wiederholt werden.
Pathophysiologie
Beim gesunden Hund münden die Harnleiter innerhalb des Trigonum vesicae (Harnblasendreieck) in die Harnblase, wo der Urin bis zur Miktion durch den Musculus urethralis zurückbehalten wird.
Bei Harnleiterektopie liegt die Mündung eines oder beider Harnleiter unterhalb dieses Schliessmuskels und mündet statt in die Harnblase in die Harnröhre, die Vagina oder den Uterus. Weil der Urin nicht zurückbehalten werden kann, kommt es zu Harninkontinenz, die sich in ständigem leichtem Harntröpfeln äußert.
Als Komplikationen können lokale Hautreizungen und aufsteigende Harnwegsinfektionen auftreten.
Klinik
Die Krankheit tritt bei Norwegischen Elchhunden, Siberian Huskies, Neufundländern, Golden und Labrador Retrievern, West Highland White Terriers, Foxterriers und Zwergpudeln gehäuft auf, kann aber bei allen Rassen sporadisch auftreten. Hündinnen sind acht mal so oft betroffen wie Rüden.
Die meisten Fälle werden zwischen dem Alter von drei und sechs Monaten vorgestellt.
Symptome
Das Leitsymptom der Harnleiterektopie ist Harninkontinenz mit ständigem Harntröpfeln.
Tiere mit einem normalen und einem ektopischen Harnleiter zeigen zusätzlich auch normales Harnabsatzverhalten, während Tiere mit beidseitig ektopischen Harnleitern nicht normal urinieren können. Durch das ständige Harntröpfeln kann es bei Hündinnen zu Hautreizungen und Infektionen im Bereich der Vagina und Vulva kommen, die bis in die Nieren aufsteigen können.
Rüden entwickeln seltener Symptome, weil sie die Umgehung des M. urethralis normalerweise durch den Musculus sphincter urethrae internus kompensieren können, der die gesamte Länge der Harnröhre umfasst.
Diagnose
Die Diagnose erfolgt über die Injektion eines Kontrastmittels in den Blutkreislauf, das über die Nieren ausgeschieden wird. Mittels Röntgenaufnahmen kann der Weg des Kontrastmittels durch Niere und Harnleiter bis in die Harnblase nachvollzogen werden.
Ist ein ektopischer Harnleiter vorhanden, zeigen die Röntgenbilder dessen Verlauf an der Blase vorbei bis zu seiner Mündung. Gleichzeitig erlaubt diese Diagnosemethode auch die Darstellung von eventuell vorhandenen zusätzlichen Anomalien im Harntrakt, wie etwa einer Hydronephrose.
Therapie und Prognose
Eine kurative Behandlung erfolgt chirurgisch.
Bei einer einseitigen Harnleiterektopie kann eine Nephrektomie der betroffenen Niere durchgeführt werden, insbesondere auch, wenn Komplikationen wie etwa eine Hydronephrose oder Pyelonephritis vorhanden sind. Bei einer beidseitigen Harnleiterektopie ist die Behandlung der Wahl eine Transplantation der ektopischen Harnleitermündungen in die Harnblase, wodurch in den Harnwegen eine funktionell normale Situation erreicht werden kann. Die Transplantation ist auch bei einseitiger Ektopie möglich, falls die Niere der betroffenen Seite normal ist.
Bei einer beidseitigen Harnleiterektopie sowie bei einer einseitigen Ektopie mit einer nicht vollkommen gesunden kontralateralen Niere ist die Nephrektomie kontraindiziert. In solchen Fällen besteht die Behandlung aus der Transplantation der ektopischen Uretermündung in die Harnblase.
In leichten Fällen kann die Harninkontinenz auch symptomatisch behandelt werden, beispielsweise durch Gaben von Phenylpropanolamin oder Ephedrin.
Bei einer erfolgreichen chirurgischen Behandlung ist die Prognose gut. Gelegentlich kommt es zu Komplikationen wie persistierender Harninkontinenz oder Dysurie sowie zu Hydronephrose.
Genetik und Zuchthygiene
Der Erbgang der Harnleiterektopie beim Hund ist nicht bekannt, vermutlich handelt es sich um eine polygene Krankheit. Betroffene Hunde sollten nicht zur Zucht verwendet werden; Verpaarungen, aus denen Welpen mit Harnleiterektopie hervorgingen, sollten nicht wiederholt werden.
eingestellt von: ise; Quellennachweis
Hound-Ataxie
Die Hound-Ataxie tritt bei Beagle und English Foxhound auf.
Als Ursache wird eine überwiegende Fütterung von Pansen diskutiert. Sie entsteht im 2.–7. Lebensjahr infolge einer Degeneration der weißen Substanz im Brustmark und der grauen Substanz im Hirnstamm und zeigt sich in Bewegungsstörungen, spastischer Parese und vermindertem Pannikulusreflex.
Als Ursache wird eine überwiegende Fütterung von Pansen diskutiert. Sie entsteht im 2.–7. Lebensjahr infolge einer Degeneration der weißen Substanz im Brustmark und der grauen Substanz im Hirnstamm und zeigt sich in Bewegungsstörungen, spastischer Parese und vermindertem Pannikulusreflex.
eingestellt von: ise; Quellennachweis
Ichthyosen in der Tiermedizin
In der Tiermedizin werden Ichthyosen besonders bei Hunden und Rindern diagnostiziert, selten auch bei anderen Haustieren. Beschrieben wurde die Krankheit bei Rindern bereits 1850. Es handelt sich dabei um eine angeborene Form (Ichthyosis congenita), die meist letal ist - betroffene Tiere werden meist bereits tot geboren.
Hunde mit leichteren Formen einer Ichthyose können symptomatisch mit Kortikosteroiden behandelt werden. Eine Heilung ist nicht möglich. Merkmalsträger sollten nicht zur Zucht verwendet werden.
Hunde mit leichteren Formen einer Ichthyose können symptomatisch mit Kortikosteroiden behandelt werden. Eine Heilung ist nicht möglich. Merkmalsträger sollten nicht zur Zucht verwendet werden.
eingestellt von: ise; Quellennachweis
Kartagener-Syndrom und PCD bei Tieren
PCD kommt gelegentlich auch bei Haushunden vor, wobei in etwa der Hälfte der Fälle auch ein Situs inversus, also ein Kartagener-Syndrom, beobachtet wird. Häufiger betroffene Hunderassen sind English Springer Spaniel, Bobtail, Shar Pei und Bichon Frisé.
Die Symptome können nur schwach ausgebildet sein und sich mit dem Alter verschlechtern. Typische Beschwerden sind anhaltender schleimig-eitriger Nasenausfluss, produktiver Husten, verminderte Leistungsfähigkeit und Dyspnoe.
Therapie
Die Therapie erfolgt symptomatisch wie beim Menschen. Die Behandlung entspricht im Wesentlichen dem Vorgehen bei chronischer Bronchitis oder Sinusitis (physikalische Therapie, Schleimlösung, Antibiotika, Polypentfernung usw.)
Die Symptome können nur schwach ausgebildet sein und sich mit dem Alter verschlechtern. Typische Beschwerden sind anhaltender schleimig-eitriger Nasenausfluss, produktiver Husten, verminderte Leistungsfähigkeit und Dyspnoe.
Therapie
Die Therapie erfolgt symptomatisch wie beim Menschen. Die Behandlung entspricht im Wesentlichen dem Vorgehen bei chronischer Bronchitis oder Sinusitis (physikalische Therapie, Schleimlösung, Antibiotika, Polypentfernung usw.)
eingestellt von: ise; Quellennachweis
Kongenitales Vestibularsyndrom
Das Kongenitale Vestibularsyndrom (auch Kongenitales Vestibulärsyndrom) ist eine seltene Erbkrankheit bei Hunden und Katzen infolge einer Fehlbildung des Gleichgewichtsorgans im Innenohr. Sie äußert sich in Kopfschiefhaltung, Koordinationsstörungen und häufig auch Taubheit.
Vorkommen und Ursachen
Das Kongenitale Vestibularsyndrom tritt bei einigen Rassen gehäuft auf. Vermutet wird ein autosomal-rezessiver Erbgang. Bei Hunden sind vor allem Akita-Inu, Cocker Spaniel, Deutscher Schäferhund, Dobermann und Tibet-Terrier betroffen.
Betroffene Tiere zeigen eine Fehlbildung des Gleichgewichtsorgans mit fehlenden oder missgebildeten Statolithen sowie häufig auch eine Degeneration der Haarzellen des Corti-Organs in der Hörschnecke.
Klinik
Die Erkrankung zeigt erste Symptome innerhalb des ersten Lebensmonats.
Dabei treten Kopfschiefhaltung und Gleichgewichtsstörungen mit Neigung zum Umfallen und leichten Bewegungsstörungen. Meist tritt gleichzeitig eine ein- oder beidseitige Taubheit auf, die durch einen Hörtest oder eine Hirnstammaudiometrie nachgewiesen werden kann.
Ein Augenzittern (Nystagmus) tritt, im Gegensatz zu den meisten anderen Gleichgewichtserkrankungen, nicht auf, allerdings lässt sich oft auch kein physiologischer Nystagmus auslösen. Die Diagnose lässt sich anhand der Rasse- und Altersprädisposition und dem klinischen Bild stellen.
Eine Behandlung ist nicht möglich. Der Krankheitsverlauf ist sehr variabel.
Zumeist stellt sich ab dem 2. Lebensmonat eine Besserung ein, da die Funktionsstörung des Gleichgewichtsorgans durch andere Sinne (Gesichtssinn, Propriozeption) ausgeglichen wird, so dass trotz Taubheit ein weitgehend beschwerdefreies Leben möglich ist. Betroffene Tiere sollten allerdings von der Zucht ausgeschlossen werden.
Vorkommen und Ursachen
Das Kongenitale Vestibularsyndrom tritt bei einigen Rassen gehäuft auf. Vermutet wird ein autosomal-rezessiver Erbgang. Bei Hunden sind vor allem Akita-Inu, Cocker Spaniel, Deutscher Schäferhund, Dobermann und Tibet-Terrier betroffen.
Betroffene Tiere zeigen eine Fehlbildung des Gleichgewichtsorgans mit fehlenden oder missgebildeten Statolithen sowie häufig auch eine Degeneration der Haarzellen des Corti-Organs in der Hörschnecke.
Klinik
Die Erkrankung zeigt erste Symptome innerhalb des ersten Lebensmonats.
Dabei treten Kopfschiefhaltung und Gleichgewichtsstörungen mit Neigung zum Umfallen und leichten Bewegungsstörungen. Meist tritt gleichzeitig eine ein- oder beidseitige Taubheit auf, die durch einen Hörtest oder eine Hirnstammaudiometrie nachgewiesen werden kann.
Ein Augenzittern (Nystagmus) tritt, im Gegensatz zu den meisten anderen Gleichgewichtserkrankungen, nicht auf, allerdings lässt sich oft auch kein physiologischer Nystagmus auslösen. Die Diagnose lässt sich anhand der Rasse- und Altersprädisposition und dem klinischen Bild stellen.
Eine Behandlung ist nicht möglich. Der Krankheitsverlauf ist sehr variabel.
Zumeist stellt sich ab dem 2. Lebensmonat eine Besserung ein, da die Funktionsstörung des Gleichgewichtsorgans durch andere Sinne (Gesichtssinn, Propriozeption) ausgeglichen wird, so dass trotz Taubheit ein weitgehend beschwerdefreies Leben möglich ist. Betroffene Tiere sollten allerdings von der Zucht ausgeschlossen werden.
eingestellt von: ise; Quellennachweis
Kraniomandibuläre Osteopathie
Die kraniomandibuläre Osteopathie (auch Cranio-mandibuläre Osteopathie, CMO, oder Osteopathia craniomandibularis hypertrophicans) ist eine erblich bedingte, sehr schmerzhafte Knochenerkrankung des Schädels bei Haushunden, vor allem bei West Highland White Terriern. Die Erkrankung wurde 1958 durch Littleworth erstmals beschrieben.
Vorkommen
Die kraniomandibuläre Osteopathie kommt vor allem bei West Highland White und bei mit ihm nahe verwandten Rassen wie Cairn und Scottish Terriern vor. Einzelfälle wurden auch für andere Rassen wie Deutscher Schäferhund, Labrador Retriever, Dobermann, Deutsche Dogge und Deutsch Drahthaar beschrieben.
Klinisches Bild
Die Erkrankung beginnt ab einem Alter von etwa 4 Monaten.
Die betroffenen Tiere zeigen eine starke Schmerzhaftigkeit der Kiefergelenksregion, Berührungen des Kopfes werden nicht mehr toleriert. Sie vermeiden es, den Fang zu öffnen und die Futteraufnahme wird, trotz erhaltenen Appetits, stark vermindert oder sogar eingestellt.
Gelegentlich können auch Fieberschübe auftreten. Die Symptome verschlechtern sich zunächst zunehmend, ab einem Alter von einem Jahr bilden sich jedoch die Knochenveränderungen wieder spontan zurück.
Röntgenuntersuchung
Röntgenologisch zeigen sich Knochenzubildungen und Sklerosierungen an Unterkiefer, Kiefergelenk oder Bulla tympanica (knöcherne Kapsel um das Mittelohr). Gelegentlich sind auch Veränderungen an den Gliedmaßenknochen nachweisbar. Osteolytische Prozesse oder Infiltrationen des umgebenden Gewebes treten nicht auf.
Zu Beginn sind die Knochenveränderungen nur gering ausgeprägt und können daher übersehen werden. Anfangs bilden sich nur flach geschichtete Zubildungen, die eine leichte Unregelmäßigkeit der Knochenkonturen von Unterkieferkörper und Bulla tympanica verursachen. Für den Nachweis solcher geringgradigen Veränderungen eignen sich vor allem Schrägaufnahmen, bei denen der Kopf gestreckt und etwa 30–40° zur Seite gedreht wird.
Im weiteren Krankheitsverlauf treten durch Auflösung der Knochenbälkchen wolkige Verschattungen und schließlich deutliche Verdickungen auf. Sie zeigen meist einen geschichteten Aufbau, der durch den schubweisen Krankheitsverlauf verursacht wird.
Differentialdiagnostisch müssen gutartige (Osteome) oder bösartige (Osteosarkome) Knochentumore sowie das Cavarial-Hyperostosis-Syndrom ausgeschlossenen werden.
Pathologie
Pathohistologisch kommt es zu einer Knochenresorption durch Osteoklasten, wobei der Lamellenknochen durch Geflechtknochen ersetzt wird. Das Knochenmark der Hohlräume der Substantia spongiosa wird durch stark durchblutetes Bindegewebe ersetzt. Am Rand der Knochenzubildungen sind Entzündungszellen zu finden.
Behandlung
Ein kausale Therapie ist nicht möglich. Ziel der Behandlung ist es, für die Zeit bis zur spontanen Remission die Schmerzen zu lindern. Hier werden zumeist Entzündungshemmer wie Carprofen, Meloxicam oder Metamizol eingesetzt. Einige Autoren setzen alternativ Prednisolon ein.
Erbgang und Zuchthygiene
Bei Scottish, Cairn und West Highland White Terrier ist die Krankheit mit einer Mutation eines Gens auf Chromosom 5 assoziiert. Der Erbgang ist bei diesen Rassen einfach dominant mit unvollständiger Penetranz. Die Penetranz beträgt bei für die Mutation reinerbigen Hunden 57 %. Für diese drei Rassen steht ein Gentest zur Verfügung. Für die anderen von kraniomandibulärer Osteopathie betroffenen Rassen existiert bislang kein Gentest.
Aufgrund der vermuteten erblichen Genese sollten betroffene Tiere nicht zur Zucht eingesetzt werden.
Verpaarungen aus denen CMO-Tiere hervorgingen sollten nicht wieder vorgenommen werden. Stur et al. (1991) empfehlen auch den Zuchtausschluss gesunder Geschwister erkrankter Tiere.
Vorkommen
Die kraniomandibuläre Osteopathie kommt vor allem bei West Highland White und bei mit ihm nahe verwandten Rassen wie Cairn und Scottish Terriern vor. Einzelfälle wurden auch für andere Rassen wie Deutscher Schäferhund, Labrador Retriever, Dobermann, Deutsche Dogge und Deutsch Drahthaar beschrieben.
Klinisches Bild
Die Erkrankung beginnt ab einem Alter von etwa 4 Monaten.
Die betroffenen Tiere zeigen eine starke Schmerzhaftigkeit der Kiefergelenksregion, Berührungen des Kopfes werden nicht mehr toleriert. Sie vermeiden es, den Fang zu öffnen und die Futteraufnahme wird, trotz erhaltenen Appetits, stark vermindert oder sogar eingestellt.
Gelegentlich können auch Fieberschübe auftreten. Die Symptome verschlechtern sich zunächst zunehmend, ab einem Alter von einem Jahr bilden sich jedoch die Knochenveränderungen wieder spontan zurück.
Röntgenuntersuchung
Röntgenologisch zeigen sich Knochenzubildungen und Sklerosierungen an Unterkiefer, Kiefergelenk oder Bulla tympanica (knöcherne Kapsel um das Mittelohr). Gelegentlich sind auch Veränderungen an den Gliedmaßenknochen nachweisbar. Osteolytische Prozesse oder Infiltrationen des umgebenden Gewebes treten nicht auf.
Zu Beginn sind die Knochenveränderungen nur gering ausgeprägt und können daher übersehen werden. Anfangs bilden sich nur flach geschichtete Zubildungen, die eine leichte Unregelmäßigkeit der Knochenkonturen von Unterkieferkörper und Bulla tympanica verursachen. Für den Nachweis solcher geringgradigen Veränderungen eignen sich vor allem Schrägaufnahmen, bei denen der Kopf gestreckt und etwa 30–40° zur Seite gedreht wird.
Im weiteren Krankheitsverlauf treten durch Auflösung der Knochenbälkchen wolkige Verschattungen und schließlich deutliche Verdickungen auf. Sie zeigen meist einen geschichteten Aufbau, der durch den schubweisen Krankheitsverlauf verursacht wird.
Differentialdiagnostisch müssen gutartige (Osteome) oder bösartige (Osteosarkome) Knochentumore sowie das Cavarial-Hyperostosis-Syndrom ausgeschlossenen werden.
Pathologie
Pathohistologisch kommt es zu einer Knochenresorption durch Osteoklasten, wobei der Lamellenknochen durch Geflechtknochen ersetzt wird. Das Knochenmark der Hohlräume der Substantia spongiosa wird durch stark durchblutetes Bindegewebe ersetzt. Am Rand der Knochenzubildungen sind Entzündungszellen zu finden.
Behandlung
Ein kausale Therapie ist nicht möglich. Ziel der Behandlung ist es, für die Zeit bis zur spontanen Remission die Schmerzen zu lindern. Hier werden zumeist Entzündungshemmer wie Carprofen, Meloxicam oder Metamizol eingesetzt. Einige Autoren setzen alternativ Prednisolon ein.
Erbgang und Zuchthygiene
Bei Scottish, Cairn und West Highland White Terrier ist die Krankheit mit einer Mutation eines Gens auf Chromosom 5 assoziiert. Der Erbgang ist bei diesen Rassen einfach dominant mit unvollständiger Penetranz. Die Penetranz beträgt bei für die Mutation reinerbigen Hunden 57 %. Für diese drei Rassen steht ein Gentest zur Verfügung. Für die anderen von kraniomandibulärer Osteopathie betroffenen Rassen existiert bislang kein Gentest.
Aufgrund der vermuteten erblichen Genese sollten betroffene Tiere nicht zur Zucht eingesetzt werden.
Verpaarungen aus denen CMO-Tiere hervorgingen sollten nicht wieder vorgenommen werden. Stur et al. (1991) empfehlen auch den Zuchtausschluss gesunder Geschwister erkrankter Tiere.
eingestellt von: ise; Quellennachweis
Kupfertoxikose
Die Kupfertoxikose ist eine Erbkrankheit des Hundes, die mit dem Morbus Wilson des Menschen vergleichbar ist. Sie wurde zuerst beim Bedlington Terrier beschrieben, kommt aber auch bei anderen Rassen vor.
Pathophysiologie
Die betroffenen Hunde leiden unter einem genetischen Stoffwechseldefekt, der verhindert, dass Kupfer in der Leber über die Galle ausgeschieden werden kann. Das Kupfer reichert sich in den Leberzellen an und führt in diesen zu einer oxidativen Schädigung der Mitochondrien.
Dadurch entsteht eine chronisch-aktive Hepatitis, die schließlich in eine Leberzirrhose übergeht. Gelegentlich kommt es auch zu einer akuten Lebernekrose mit Freisetzung von Kupfer in die Blutbahn und dadurch verursachter Hämolyse.
Klinik
Symptome können prinzipiell in jedem Alter auftreten; vorgestellt werden aber meist mittelalte Hunde der Rassen Bedlington Terrier, Skye Terrier, West Highland White Terrier und Dobermann, wobei die Erkrankung beim Bedlington Terrier proportional am häufigsten auftritt. Fälle sind auch bei anderen Rassen beschrieben, darunter Dalmatiner, Keeshond und Labrador Retriever.
Symptome
Die Symptome variieren je nach Krankheitsverlauf. Bei der chronischen Form steht langsam fortschreitender Leistungsverlust und Abmagerung im Vordergrund, daneben kann Ikterus vorhanden sein. Typisch ist eine verkleinerte Leber; auch hepatische Enzephalopathie kann auftreten. Bei akuter Lebernekrose mit Hämolyse kommt es akut zu Erbrechen, Anorexie und Depression.
Diagnose
Neben den üblichen Zeichen für ein akutes oder chronisches Leberproblem wird die Diagnose durch eine Leberbiopsie gestellt, bei der eine stark erhöhte Kupferkonzentration im Gewebe festgestellt werden kann. Für den Bedlington Terrier existiert ein DNA-Markertest, der ebenfalls zur Diagnose geeignet ist.
Therapie und Prognose
Die Therapie ist symptomatisch und besteht in einer geeigneten Diät in Verbindung mit Chelation des Kupfers, die eine Ausscheidung über die Nieren ermöglicht. Daneben werden Hepatitis und Zirrhose je nach Schweregrad ebenfalls behandelt.
Die Prognose bei akuter Lebernekrose und Hämolyse sowie bei Hunden mit Leberzirrhose ist schlecht. Hunde mit leichtem bis mittelgradigem Leberversagen haben eine mäßig gute Prognose. Bei einer Diagnose vor dem Auftreten von Hepatitis ist die Prognose gut.
Genetik und Zuchthygiene
Die Krankheit ist beim Bedlington Terrier als einfach autosomal rezessiv beschrieben, wobei wie erwähnt ein DNA-Test zur Verfügung steht, mittels dessen auch klinisch gesunde Träger erkannt werden können.
Es ist unklar, inwieweit dieser Test auch auf andere Rassen anwendbar ist. Wird bei einem nicht-Bedlington-Hund Kupfertoxikose diagnostiziert, ist zu empfehlen, seine Verwandten einer Leberbiopsie zu unterziehen, um bei diesen gegebenenfalls präventive Maßnahmen einleiten zu können.
Ohne DNA-Test wird empfohlen, Eltern, Nachkommen der ersten Generation und Vollgeschwister eines Kupfertoxikose-Hundes von der Zucht auszuschließen.
Pathophysiologie
Die betroffenen Hunde leiden unter einem genetischen Stoffwechseldefekt, der verhindert, dass Kupfer in der Leber über die Galle ausgeschieden werden kann. Das Kupfer reichert sich in den Leberzellen an und führt in diesen zu einer oxidativen Schädigung der Mitochondrien.
Dadurch entsteht eine chronisch-aktive Hepatitis, die schließlich in eine Leberzirrhose übergeht. Gelegentlich kommt es auch zu einer akuten Lebernekrose mit Freisetzung von Kupfer in die Blutbahn und dadurch verursachter Hämolyse.
Klinik
Symptome können prinzipiell in jedem Alter auftreten; vorgestellt werden aber meist mittelalte Hunde der Rassen Bedlington Terrier, Skye Terrier, West Highland White Terrier und Dobermann, wobei die Erkrankung beim Bedlington Terrier proportional am häufigsten auftritt. Fälle sind auch bei anderen Rassen beschrieben, darunter Dalmatiner, Keeshond und Labrador Retriever.
Symptome
Die Symptome variieren je nach Krankheitsverlauf. Bei der chronischen Form steht langsam fortschreitender Leistungsverlust und Abmagerung im Vordergrund, daneben kann Ikterus vorhanden sein. Typisch ist eine verkleinerte Leber; auch hepatische Enzephalopathie kann auftreten. Bei akuter Lebernekrose mit Hämolyse kommt es akut zu Erbrechen, Anorexie und Depression.
Diagnose
Neben den üblichen Zeichen für ein akutes oder chronisches Leberproblem wird die Diagnose durch eine Leberbiopsie gestellt, bei der eine stark erhöhte Kupferkonzentration im Gewebe festgestellt werden kann. Für den Bedlington Terrier existiert ein DNA-Markertest, der ebenfalls zur Diagnose geeignet ist.
Therapie und Prognose
Die Therapie ist symptomatisch und besteht in einer geeigneten Diät in Verbindung mit Chelation des Kupfers, die eine Ausscheidung über die Nieren ermöglicht. Daneben werden Hepatitis und Zirrhose je nach Schweregrad ebenfalls behandelt.
Die Prognose bei akuter Lebernekrose und Hämolyse sowie bei Hunden mit Leberzirrhose ist schlecht. Hunde mit leichtem bis mittelgradigem Leberversagen haben eine mäßig gute Prognose. Bei einer Diagnose vor dem Auftreten von Hepatitis ist die Prognose gut.
Genetik und Zuchthygiene
Die Krankheit ist beim Bedlington Terrier als einfach autosomal rezessiv beschrieben, wobei wie erwähnt ein DNA-Test zur Verfügung steht, mittels dessen auch klinisch gesunde Träger erkannt werden können.
Es ist unklar, inwieweit dieser Test auch auf andere Rassen anwendbar ist. Wird bei einem nicht-Bedlington-Hund Kupfertoxikose diagnostiziert, ist zu empfehlen, seine Verwandten einer Leberbiopsie zu unterziehen, um bei diesen gegebenenfalls präventive Maßnahmen einleiten zu können.
Ohne DNA-Test wird empfohlen, Eltern, Nachkommen der ersten Generation und Vollgeschwister eines Kupfertoxikose-Hundes von der Zucht auszuschließen.
eingestellt von: ise; Quellennachweis
Leukoenzephalomyelopathie des Rottweilers
Die Leukoenzephalomyelopathie des Rottweilers ist eine vermutlich erblich bedingte Demyelinisierung des gesamten Zentralnervensystems, am stärksten im Rückenmark und Hirnstamm, bei Rottweilern.
Sie entwickelt sich innerhalb der ersten drei Lebensjahre als fortschreitende Ataxie. Die Rückenmarksreflexe bleiben erhalten. Innerhalb eines halben Jahres können betroffene Tiere nicht mehr aufstehen und stehen.
Sie entwickelt sich innerhalb der ersten drei Lebensjahre als fortschreitende Ataxie. Die Rückenmarksreflexe bleiben erhalten. Innerhalb eines halben Jahres können betroffene Tiere nicht mehr aufstehen und stehen.
eingestellt von: ise; Quellennachweis
Lundehundsyndrom

Das Lundehundsyndrom/Lundehund-Gastroenteropathie ist ein Komplex von Krankheiten des Verdauungstrakts, der bei Norwegischen Lundehunden auftritt.
Aufgrund des gehäuften Auftretens der Erkrankung bei dieser Hunderasse ist davon auszugehen, dass es sich um eine Erbkrankheit handelt.
Pathophysiologie
Das Lundehundsyndrom gehört zum Komplex der chronisch-entzündlichen Darmerkrankungen (IBD) und umfasst Gastritis, Proteinverlust über den Darm (Protein Losing Enteropathy, PLE), Lymphangiektasie und Malabsorption.
Klinik
Das Lundehundsyndrom tritt ausschließlich bei Norwegischen Lundehunden auf.
Am häufigsten wird es in mittlerem Alter (ca. fünf Jahre) diagnostiziert, kann jedoch prinzipiell in jedem Lebensalter auftreten. Eine Geschlechtsdisposition besteht nicht, Rüden und Hündinnen scheinen gleich häufig betroffen zu sein. Die typischen Veränderungen des Magen-Darm-Trakts finden sich häufig auch bei klinisch nicht oder noch nicht erkrankten Lundehunden.
Symptome
Betroffene Hunde können verschiedene Magen-Darm-Symptome entwickeln, wie etwa intermittierenden Durchfall, Gastritis, Erbrechen, Gewichtsabnahme und Bauchwassersucht.
Daneben kann es aufgrund von Proteinverlusten durch den Darm auch zu einem Ödem der Unterhaut (Anasarka) kommen, das besonders an den Hinterbeinen auftritt. In schweren Fällen können auch Hydrothorax und Hydroperikard auftreten.
Diagnose
Die Diagnose erfolgt über eine mikroskopische Untersuchung von Gewebeproben der Magen-Darm-Schleimhaut, die bei der Endoskopie entnommen werden. Makroskopisch findet man ein Ödem und partielle Erosionen der Magenschleimhaut sowie eine durch Fibrose verdickte Magenwand. Mikroskopisch ist eine chronisch-atrophische Gastritis mit mononukleärer Infiltration und Degeneration der Fundusdrüsen feststellbar.
Bei manchen Hunden finden sich daneben auch erweiterte Lymphgefäße (Lymphangiektasie). In der Nähe des Magenpförtners können sich entzündliche Knoten entwickeln. Im Gegensatz zu den chronisch-entzündlichen Darmerkrankungen des Menschen scheint das Lundehundsyndrom nicht zu einer Metaplasie der Darmepithelzellen zu führen. Im Dünndarm finden sich öfters ebenfalls erweiterte Lymphgefäße. Die Darmschleimhaut ist verdickt und rauh. Die Zotten sind verkümmert und zum Teil miteinander verschmolzen, die Krypten dagegen hyperplastisch und zum Teil mit Zellfragmenten gefüllt. Teilweise kommt es zur Ablösung des Epithels und Blasenbildung zwischen Basalmembran und Epithel. Auch Granulome können auftreten.
Das Lundehundsyndrom verursacht keine charakteristischen Veränderungen des Blutbilds.
Im Gegensatz zu anderen von PLE betroffenen Rassen sind die Lymphozytenwerte beim Lundehundsyndrom normal. Totalprotein, Albumin, Calcium, Cobalamin und Folsäure können vermindert sein.
Als zusätzlichen diagnostischen Test gibt es auch die Möglichkeit, den Kot des Hundes auf fäkalen α1-Proteinase-Inhibitor zu untersuchen. Ein erhöhter Wert deutet auf einen Proteinverlust über den Darm hin, der bei Lundehunden als diagnostisch für Lundehundsyndrom gelten kann.
Therapie und Prognose
Die Behandlung des Lundehundsyndroms ist rein symptomatisch, da die Ursache der Magen-Darm-Veränderungen bisher unbekannt ist. Es gibt kein Therapieprotokoll, das bei allen Hunden zur Anwendung kommen kann, und eine Verbesserung der Symptome unter Behandlung ist individuell sehr unterschiedlich.
Generell wird eine qualitativ hochwertige, leicht verdauliche Diät mit niedrigem Fett- und hohem Proteingehalt empfohlen, um das Lymphsystem des Darms zu entlasten. Gelegentlich wird die Zugabe von mittellangen Triglyceriden empfohlen, die auch über eine nicht-lymphatische Route aufgenommen werden können. Vitamingaben können ebenfalls sinnvoll sein, besonders in Fällen, wo Cobalamin und Folsäure erniedrigt sind; Cobalamin muss dabei parenteral verabreicht werden. Entzündungshemmer und Immunsuppressiva wie etwa Prednisolon können die Symptome ebenfalls verbessern, wobei Prednisolon auch die Fähigkeit des Darm zu Absorption von Nährstoffen zu verbessern scheint. Bei häufigem Erbrechen können auch Antiemetika gegeben werden. Die Behandlung ist lebenslänglich, eine Heilung ist nicht möglich.
Komplikationen des Lundehundsyndroms sind eine bakterielle Besiedlung des Dünndarms (SIBO), die durch die Gabe von Antibiotika behandelt werden kann. Ödembildung wird durch Diuretika behandelt. Durch den Verlust von Antithrombin III durch den Darm kann es zu einer erhöhten Blutgerinnungsneigung kommen, die zu Thrombosen und Embolien führen kann. Die chronische Gastritis kann zu einem bösartigen Tumor der Magenwand entarten (Magenkarzinom).
Die Prognose ist sehr unterschiedlich und hängt davon ab, wie der Hund auf die Behandlung reagiert. Die Erkrankung verläuft aber auch bei behandelten Hunden oft fortschreitend und kann schließlich zum Tod führen. Manche Lundehunde müssen nur bei akuten Phasen behandelt werden und haben dazwischen lange symptomfreie Intervalle, während andere sich nie mehr von den ersten Symptomen erholen.
Genetik und Zuchthygiene
Die heutige Lundehundpopulation geht auf fünf Individuen zurück, mit denen im Jahr 1961 mit einer kontrollierten Zucht begonnen wurde. Einer dieser Hunde entwickelte im Alter von drei Jahren Symptome, die denen des Lundehundsyndroms entsprechen.
Seither wurde die Erkrankung immer wieder bei Hunden der Rasse beobachtet. Man kann daher davon ausgehen, dass es sich um eine Erbkrankheit handelt, die sich durch einen genetischen Flaschenhals innerhalb der ganzen Rasse ausbreiten konnte (Gründereffekt). Die Prävalenz der Krankheit innerhalb der Rasse ist sehr hoch, bei etwa der Hälfte der untersuchten Lundehunde konnte ein Proteinverlust über den Darm nachgewiesen werden.
Insgesamt kann aufgrund der Komplexität des Syndroms von einer polygenen Genese ausgegangen werden. Somit wäre eine Zuchtwertschätzung zur Bekämpfung der Krankheit empfehlenswert, jedoch aufgrund der hohen Prävalenz schwierig.
Aufgrund des gehäuften Auftretens der Erkrankung bei dieser Hunderasse ist davon auszugehen, dass es sich um eine Erbkrankheit handelt.
Pathophysiologie
Das Lundehundsyndrom gehört zum Komplex der chronisch-entzündlichen Darmerkrankungen (IBD) und umfasst Gastritis, Proteinverlust über den Darm (Protein Losing Enteropathy, PLE), Lymphangiektasie und Malabsorption.
Klinik
Das Lundehundsyndrom tritt ausschließlich bei Norwegischen Lundehunden auf.
Am häufigsten wird es in mittlerem Alter (ca. fünf Jahre) diagnostiziert, kann jedoch prinzipiell in jedem Lebensalter auftreten. Eine Geschlechtsdisposition besteht nicht, Rüden und Hündinnen scheinen gleich häufig betroffen zu sein. Die typischen Veränderungen des Magen-Darm-Trakts finden sich häufig auch bei klinisch nicht oder noch nicht erkrankten Lundehunden.
Symptome
Betroffene Hunde können verschiedene Magen-Darm-Symptome entwickeln, wie etwa intermittierenden Durchfall, Gastritis, Erbrechen, Gewichtsabnahme und Bauchwassersucht.
Daneben kann es aufgrund von Proteinverlusten durch den Darm auch zu einem Ödem der Unterhaut (Anasarka) kommen, das besonders an den Hinterbeinen auftritt. In schweren Fällen können auch Hydrothorax und Hydroperikard auftreten.
Diagnose
Die Diagnose erfolgt über eine mikroskopische Untersuchung von Gewebeproben der Magen-Darm-Schleimhaut, die bei der Endoskopie entnommen werden. Makroskopisch findet man ein Ödem und partielle Erosionen der Magenschleimhaut sowie eine durch Fibrose verdickte Magenwand. Mikroskopisch ist eine chronisch-atrophische Gastritis mit mononukleärer Infiltration und Degeneration der Fundusdrüsen feststellbar.
Bei manchen Hunden finden sich daneben auch erweiterte Lymphgefäße (Lymphangiektasie). In der Nähe des Magenpförtners können sich entzündliche Knoten entwickeln. Im Gegensatz zu den chronisch-entzündlichen Darmerkrankungen des Menschen scheint das Lundehundsyndrom nicht zu einer Metaplasie der Darmepithelzellen zu führen. Im Dünndarm finden sich öfters ebenfalls erweiterte Lymphgefäße. Die Darmschleimhaut ist verdickt und rauh. Die Zotten sind verkümmert und zum Teil miteinander verschmolzen, die Krypten dagegen hyperplastisch und zum Teil mit Zellfragmenten gefüllt. Teilweise kommt es zur Ablösung des Epithels und Blasenbildung zwischen Basalmembran und Epithel. Auch Granulome können auftreten.
Das Lundehundsyndrom verursacht keine charakteristischen Veränderungen des Blutbilds.
Im Gegensatz zu anderen von PLE betroffenen Rassen sind die Lymphozytenwerte beim Lundehundsyndrom normal. Totalprotein, Albumin, Calcium, Cobalamin und Folsäure können vermindert sein.
Als zusätzlichen diagnostischen Test gibt es auch die Möglichkeit, den Kot des Hundes auf fäkalen α1-Proteinase-Inhibitor zu untersuchen. Ein erhöhter Wert deutet auf einen Proteinverlust über den Darm hin, der bei Lundehunden als diagnostisch für Lundehundsyndrom gelten kann.
Therapie und Prognose
Die Behandlung des Lundehundsyndroms ist rein symptomatisch, da die Ursache der Magen-Darm-Veränderungen bisher unbekannt ist. Es gibt kein Therapieprotokoll, das bei allen Hunden zur Anwendung kommen kann, und eine Verbesserung der Symptome unter Behandlung ist individuell sehr unterschiedlich.
Generell wird eine qualitativ hochwertige, leicht verdauliche Diät mit niedrigem Fett- und hohem Proteingehalt empfohlen, um das Lymphsystem des Darms zu entlasten. Gelegentlich wird die Zugabe von mittellangen Triglyceriden empfohlen, die auch über eine nicht-lymphatische Route aufgenommen werden können. Vitamingaben können ebenfalls sinnvoll sein, besonders in Fällen, wo Cobalamin und Folsäure erniedrigt sind; Cobalamin muss dabei parenteral verabreicht werden. Entzündungshemmer und Immunsuppressiva wie etwa Prednisolon können die Symptome ebenfalls verbessern, wobei Prednisolon auch die Fähigkeit des Darm zu Absorption von Nährstoffen zu verbessern scheint. Bei häufigem Erbrechen können auch Antiemetika gegeben werden. Die Behandlung ist lebenslänglich, eine Heilung ist nicht möglich.
Komplikationen des Lundehundsyndroms sind eine bakterielle Besiedlung des Dünndarms (SIBO), die durch die Gabe von Antibiotika behandelt werden kann. Ödembildung wird durch Diuretika behandelt. Durch den Verlust von Antithrombin III durch den Darm kann es zu einer erhöhten Blutgerinnungsneigung kommen, die zu Thrombosen und Embolien führen kann. Die chronische Gastritis kann zu einem bösartigen Tumor der Magenwand entarten (Magenkarzinom).
Die Prognose ist sehr unterschiedlich und hängt davon ab, wie der Hund auf die Behandlung reagiert. Die Erkrankung verläuft aber auch bei behandelten Hunden oft fortschreitend und kann schließlich zum Tod führen. Manche Lundehunde müssen nur bei akuten Phasen behandelt werden und haben dazwischen lange symptomfreie Intervalle, während andere sich nie mehr von den ersten Symptomen erholen.
Genetik und Zuchthygiene
Die heutige Lundehundpopulation geht auf fünf Individuen zurück, mit denen im Jahr 1961 mit einer kontrollierten Zucht begonnen wurde. Einer dieser Hunde entwickelte im Alter von drei Jahren Symptome, die denen des Lundehundsyndroms entsprechen.
Seither wurde die Erkrankung immer wieder bei Hunden der Rasse beobachtet. Man kann daher davon ausgehen, dass es sich um eine Erbkrankheit handelt, die sich durch einen genetischen Flaschenhals innerhalb der ganzen Rasse ausbreiten konnte (Gründereffekt). Die Prävalenz der Krankheit innerhalb der Rasse ist sehr hoch, bei etwa der Hälfte der untersuchten Lundehunde konnte ein Proteinverlust über den Darm nachgewiesen werden.
Insgesamt kann aufgrund der Komplexität des Syndroms von einer polygenen Genese ausgegangen werden. Somit wäre eine Zuchtwertschätzung zur Bekämpfung der Krankheit empfehlenswert, jedoch aufgrund der hohen Prävalenz schwierig.
eingestellt von: ise; Quellen- und Bildnachweis
x

Quellen- und Bildnachweis
Bildname:
Lundehundsyndrom
Urheber:
Karen Elise Dahlmo/GFDL, cc-by-sa-2.5
Link:
Maligne Histiozytose

Die Maligne Histiozytose oder das Histiozytäre Sarkom ist eine Krebserkrankung des Hundes, die vor allem beim Berner Sennenhund vorkommt und bei dieser Rasse als Erbkrankheit eine der häufigsten Todesursachen darstellt.
Bei anderen Hunderassen kommt die Erkrankung nur selten vor. Die Krankheit Maligne Histiozytose kann als Hautform („kutane Histiozytose“) oder als generalisierte Form („disseminierte Histiozytose“) auftreten.
Pathophysiologie
Histiozyten sind besonders im Bindegewebe vorkommende Zellen, die zu den Makrophagen gehören und im Gewebe gewisse Funktionen des Immunsystems wahrnehmen. Durch Mutation können diese Zellen zu bösartigen Zellen entarten und so einen Tumor bilden, der eine ausgeprägte Neigung zur Metastasenbildung besitzt.
Diese Metastasen zerstören durch ihr unkontrolliertes und invasives Wachstum lebenswichtige Organe, was schliesslich zum Tod des Hundes führt.
Klinik
An maligner Histiozytose erkranken typischerweise junge bis mittelalte Hunde zwischen dem Alter von zwei bis acht Jahren. Rüden sind häufiger betroffen als Hündinnen.
Die überwiegende Mehrzahl der Fälle tritt beim Berner Sennenhund auf. Sporadische Fälle können grundsätzlich bei allen Rassen auf, Golden Rtreten, wobei eine gewisse Häufung beim Flat Coated Teriever, Golden Retriever, Irischen Wolfshund und Rottweiler beschrieben ist.
Symptome
Von der Hautform betroffene Hunde zeigen Knoten- und Plaquebildung in der Haut – bei Rüden besonders am Hodensack – sowie der Nasen- und Augenschleimhaut. Die Läsionen sind schlecht begrenzt, gelegentlich von Alopezie begleitet und können ulzerieren. Sie entwickeln sich in Schüben und können sich nach einem Schub auch wieder langsam zurückbilden, um nach einigen Monaten erneut zu erscheinen. Die Erkrankung verschlimmert sich in der Regel mit jedem neuen Schub. Die Läsionen können sich auch auf andere Organe ausbreiten, besonders Lymphknoten, Milz und Knochenmark.
Die disseminierte Maligne Histiozytose verläuft normalerweise ohne Beteiligung der Haut. Am häufigsten betroffen sind Lunge, Lymphknoten und Leber. Betroffene Hunde zeigen Schwäche, Appetitlosigkeit, Abgeschlagenheit, Gewichtsverlust und Atemprobleme; seltener Probleme beim Schlucken oder Husten. Die Tumoren erscheinen als grosse, einzelne, feste Raumforderungen, die die befallenen Organe durch ihr invasives Wachstum zerstören.
Die disseminierte Form verläuft nicht in Schüben, sondern rasch progressiv und führt in der überwiegenden Mehrzahl der Fälle in weniger als sechs Monaten zum Tod.
Diagnose
Ein Verdacht auf kutane Histiozytose ergibt sich durch das typische Signalement des Hundes in Verbindung mit den Hauptsymptomen. Die disseminierte Histiozytose zeigt sich oft schon im Röntgenbild, wo besonders Lungenmetastasen und Knochenmetastasen zu erkennen sind.
Die Sicherung der Diagnose erfolgt durch eine mikroskopische Untersuchung von Biopsien, die aus verdächtigen Gewebsmassen entnommen werden.
Therapie und Prognose
Es existiert keine befriedigende Behandlung der Malignen Histiozytose. Chirurgisches Ausschneiden ist selten praktikabel, und die Tumoren reagieren wenig empfindlich auf Bestrahlung und Chemotherapie. Die Behandlung erfolgt daher in den meisten Fällen palliativ. Bovines Thymosin 5 zeigte bei der Behandlung der Hautform einen beschränkten Erfolg.
Die Lebenserwartung nach der Diagnose beträgt wenige Monate, nur wenige Hunde überleben länger als sechs Monate.
Genetik und Zuchthygiene
Die Maligne Histiozytose folgt beim Berner Sennenhund einem polygenen Erbgang.
Bei dieser Rasse haben verschiedene Rasseclubs zur Bekämpfung der Krankheit eine Zuchtwertschätzung eingeführt.
Bei anderen Hunderassen kommt die Erkrankung nur selten vor. Die Krankheit Maligne Histiozytose kann als Hautform („kutane Histiozytose“) oder als generalisierte Form („disseminierte Histiozytose“) auftreten.
Pathophysiologie
Histiozyten sind besonders im Bindegewebe vorkommende Zellen, die zu den Makrophagen gehören und im Gewebe gewisse Funktionen des Immunsystems wahrnehmen. Durch Mutation können diese Zellen zu bösartigen Zellen entarten und so einen Tumor bilden, der eine ausgeprägte Neigung zur Metastasenbildung besitzt.
Diese Metastasen zerstören durch ihr unkontrolliertes und invasives Wachstum lebenswichtige Organe, was schliesslich zum Tod des Hundes führt.
Klinik
An maligner Histiozytose erkranken typischerweise junge bis mittelalte Hunde zwischen dem Alter von zwei bis acht Jahren. Rüden sind häufiger betroffen als Hündinnen.
Die überwiegende Mehrzahl der Fälle tritt beim Berner Sennenhund auf. Sporadische Fälle können grundsätzlich bei allen Rassen auf, Golden Rtreten, wobei eine gewisse Häufung beim Flat Coated Teriever, Golden Retriever, Irischen Wolfshund und Rottweiler beschrieben ist.
Symptome
Von der Hautform betroffene Hunde zeigen Knoten- und Plaquebildung in der Haut – bei Rüden besonders am Hodensack – sowie der Nasen- und Augenschleimhaut. Die Läsionen sind schlecht begrenzt, gelegentlich von Alopezie begleitet und können ulzerieren. Sie entwickeln sich in Schüben und können sich nach einem Schub auch wieder langsam zurückbilden, um nach einigen Monaten erneut zu erscheinen. Die Erkrankung verschlimmert sich in der Regel mit jedem neuen Schub. Die Läsionen können sich auch auf andere Organe ausbreiten, besonders Lymphknoten, Milz und Knochenmark.
Die disseminierte Maligne Histiozytose verläuft normalerweise ohne Beteiligung der Haut. Am häufigsten betroffen sind Lunge, Lymphknoten und Leber. Betroffene Hunde zeigen Schwäche, Appetitlosigkeit, Abgeschlagenheit, Gewichtsverlust und Atemprobleme; seltener Probleme beim Schlucken oder Husten. Die Tumoren erscheinen als grosse, einzelne, feste Raumforderungen, die die befallenen Organe durch ihr invasives Wachstum zerstören.
Die disseminierte Form verläuft nicht in Schüben, sondern rasch progressiv und führt in der überwiegenden Mehrzahl der Fälle in weniger als sechs Monaten zum Tod.
Diagnose
Ein Verdacht auf kutane Histiozytose ergibt sich durch das typische Signalement des Hundes in Verbindung mit den Hauptsymptomen. Die disseminierte Histiozytose zeigt sich oft schon im Röntgenbild, wo besonders Lungenmetastasen und Knochenmetastasen zu erkennen sind.
Die Sicherung der Diagnose erfolgt durch eine mikroskopische Untersuchung von Biopsien, die aus verdächtigen Gewebsmassen entnommen werden.
Therapie und Prognose
Es existiert keine befriedigende Behandlung der Malignen Histiozytose. Chirurgisches Ausschneiden ist selten praktikabel, und die Tumoren reagieren wenig empfindlich auf Bestrahlung und Chemotherapie. Die Behandlung erfolgt daher in den meisten Fällen palliativ. Bovines Thymosin 5 zeigte bei der Behandlung der Hautform einen beschränkten Erfolg.
Die Lebenserwartung nach der Diagnose beträgt wenige Monate, nur wenige Hunde überleben länger als sechs Monate.
Genetik und Zuchthygiene
Die Maligne Histiozytose folgt beim Berner Sennenhund einem polygenen Erbgang.
Bei dieser Rasse haben verschiedene Rasseclubs zur Bekämpfung der Krankheit eine Zuchtwertschätzung eingeführt.
eingestellt von: ise; Quellen- und Bildnachweis
x
Quellen- und Bildnachweis
Bildname:
Maligne Histiozytose beim Berner Sennenhund
Urheber:
Wikipedia/Ocmey http://fr.wikipedia.org/wiki/Utilisateur:Ocmey
Link:
MDR1-Defekt
Der MDR1-Defekt ist ein Defekt im MDR1-Gen, der bei einigen Hunderassen verbreitet ist.
Dadurch kommt es zu einer mangelhaften oder fehlenden Synthese eines bestimmten Proteins (P-Glykoprotein, P-gp), welches ein wichtiger Bestandteil der Blut-Hirn-Schranke ist, was zu einer Überempfindlichkeit gegenüber manchen Arzneimitteln führt. Urheber dieses Defektes ist wahrscheinlich ein einziger Hund, der etwa Mitte des 19. Jahrhunderts gelebt hat und maßgeblich an der Entstehung und Festigung der Rasse Collie beteiligt war. Daher lässt sich dieser Defekt bei Hunderassen finden, die nachweisbar mit dem Collie verwandt sind.
Bei anderen vom Defekt betroffenen Rassen dient diese Mutation dann als Nachweis der Verwandtschaft. Ein funktionierendes MDR1-System ist vor allem bei Säugetieren (und Menschen) bekannt und hier evolutionsgeschichtlich sehr alt. Tiere, die dieses System nicht besitzen, können ähnliche Empfindlichkeiten für Medikamente zeigen.
Betroffene Rassen
Die Projektgruppe MDR1-Defekt beim Collie an der Justus-Liebig-Universität Gießen hat im Jahr 2004 im Rahmen einer Studie zur Häufigkeit des MDR1-Defektes bei verschiedenen Hunderassen Hunde aus 30 verschiedenen Rassen und 10 Europäischen Ländern getestet.
Der Defekt im MDR1-Gen wurde u. a. bei folgenden Hunderassen gefunden: Collie (Kurzhaarcollie und Langhaarcollie), Shetland Sheepdog, Australian Shepherd, Bobtail und Border-Collie. Obwohl bisher noch keine nt230(del4) Mutation beim Bearded Collie nachgewiesen werden konnte, lässt die geringe Probenzahl noch keine endgültige Bewertung zu. Bei den Rassen Wäller und Bobtail konnten bisher nur heterozygot von diesem Defekt betroffene Hunde (MDR1+/-) nachgewiesen werden, mit dem Auftreten homozygoter Mutationen müsste allerdings auch bei diesen Rassen gerechnet werden.
Beim Wäller ist es jedoch so, dass die Zuchttiere getestet sein müssen und bei der Verpaarung darauf geachtet wird, ein Trägertier nur mit einem freien zu verpaaren, damit es eben nicht zu homozygoten Welpen kommt.
Des Weiteren ist der Defekt bei folgenden Rassen bekannt: Deutscher Schäferhund, English Shepherd, McNab, Silken Windhound, Langhaarwhippet, Berger Blanc Suisse sowie einigen seltenen Rassen.
Seit Anfang 2008 werden außerdem weitere Rassen wie Barsoi, Belgischer Schäferhund oder Kelpie auf den Defekt untersucht.
Symptome, Diagnostik und Folgen für das Tier
Der Defekt im MDR1-Gen führt zu einer mangelhaften oder fehlenden Synthese des P-Glycoproteins. Dieses Eiweiß spielt eine Rolle bei ATP-abhängigen Transportvorgängen zwischen Blut und Gewebe und ist im Gehirn, in Leber, Nieren, Darm, Plazenta und Hoden zu finden. Neben seiner Funktion beim Transport körperfremder Stoffe, limitiert es den Transport der Hormone der Nebennierenrinde (Cortisol, Corticosteron) in das Gehirn und hat damit Einfluss auf die Hypothalamus-Hypophyse-Nebennierenrinden-Achse. Bei einem MDR1-Defekt kommt es daher zu einem erhöhten Übergang der Nebennierenrindenhormone in die übergeordneten Zentren und aufgrund des negativen Feedbacks zu erniedrigten Kortisolwerten im Blut.
Als eine weitere Folge des Defektes wird auch eine höhere Anfälligkeit für chronisch entzündliche Darmerkrankungen vermutet.
Bei nicht vom Defekt betroffenen Tieren dient dieses Protein u. a. dazu, körperfremde Stoffe wie Arzneimittel aus dem Körper herauszutransportieren. Es besteht also eine Art Resistenz gegenüber unerwünschten Nebenwirkungen – die sogenannte Multiple Drug Resistance.
Bekannt sind bisher die Auswirkungen auf die Blut-Hirn-Schranke. Bei dieser Grenze zwischen den Hirnblutgefässen und dem Hirnnervengewebe stellt ein sogenannter MDR1-Transporter eine Schutzbarriere für das Gehirn dar. Dieser Transporter ist Teil der Blut-Hirn-Schranke und befindet sich normalerweise auf der Oberfläche der Endothelzellen (Zellen, die die Wände der Blutgefäße auskleiden). Er sorgt dafür, dass toxische Verbindungen und Arzneistoffe in den Gehirnkapillaren zurückgehalten werden und nicht in das Gehirn eindringen können.
Besteht nun bei einem Hund der MDR1-Defekt, fehlt der Transporter und der Schutz funktioniert nicht mehr. Bei betroffenen Tieren können daher nach der Verabreichung von bestimmten Antiparasitika, Zytostatika, Durchfallmitteln oder Antibiotika starke neurotoxische Nebenwirkungen auftreten − bis zum Tod. Bei Mäusen, bei welchen der MDR1-Transporter bewusst ausgeschaltet wurde, traten nicht nur Ivermectin, sondern auch zahlreiche andere Arzneistoffe bis zu 90-fach mehr ins Gehirn als bei Vergleichstieren mit intakter Blut-Hirn-Schranke. Diese Stoffe sind auch eine potentielle Gefahr für einen vom MDR1-Defekt betroffenen Hund.
Mögliche Folge dieses Gendefektes ist die Überempfindlichkeit des Hundes gegenüber bestimmten Arzneistoffen. Da die Gabe einiger dieser Mittel bei betroffenen Hunden allerdings zum Tode führen kann, wird ein Gentest aller Hunde betroffener Rassen empfohlen. Die Universität Gießen bietet einen Test auf den MDR1-Defekt an. Zu diesem Zweck nimmt der Tierarzt dem zu untersuchenden Hund eine kleine Menge Blut ab (1 ml EDTA-Blut) und sendet diese Probe an die Universität. Mit Hilfe eines genetischen Tests wird die Blutprobe dann auf ein Vorliegen der MDR1-Mutation untersucht, dass Ergebnis wird dem Hundehalter mitgeteilt.
Ist der Hund vom Defekt betroffen, hat der Hundehalter einige Dinge zu beachten. So dürfen beispielsweise bestimmte Wurmkuren und Flohschutzmittel nicht mehr verabreicht werden. Auch bei Durchfall oder Herzerkrankungen eingesetzte Medikamente können weitreichende unerwünschte Nebenwirkungen haben. Bekannt ist eine Überempfindlichkeit z. B. für die Wirkstoffe Ivermectin, Doramectin, Moxidectin (nur bei oraler Anwendung) und Loperamid. Milbemycinoxim und Emodepsid dürfen nur unter exakter Dosierung eingesetzt werden. Viele weitere Wirkstoffe stehen aber in dem Verdacht, unerwünschte Nebenwirkungen hervorrufen zu können. Generell sollte daher der behandelnde Tierarzt über den Defekt informiert werden. Der Hund selber gilt als Risikopatient.
Bei Spaziergängen ist dann darauf zu achten, dass der Hund keinen Kot von beispielsweise Pferden zu sich nimmt, da dieser einen der gefährlichen Wirkstoffe in unveränderter Form enthalten kann.
Bisher sind Überempfindlichkeiten vor allen bei Hunden mit homozygoter Vererbung des MDR1-Defektes (MDR1 -/-) bekannt. Inzwischen wurden aber auch Reaktionen bei Trägern (MDR1 +/-) beobachtet. In einer US-amerikanischen Studie (siehe unter Links) werden die Träger deshalb als „sensitive“ bezeichnet, die vom Defekt betroffenen Hunde (MDR1 -/-) als „super sensitive“. Ebenfalls in den USA wurden die vom Defekt betroffenen Hunde inzwischen von der Forschung als Versuchstiere entdeckt.
Auswirkungen auf die Zucht
Aufgrund der Probleme in der Arzneitherapie von Hunden mit dem Genotyp MDR1(-/-) wird z. B. von der Universität in Gießen empfohlen, den Gendefekt in der Zucht betroffener Hunderassen zu berücksichtigen und so zu verpaaren, dass keine vom Defekt betroffenen Nachkommen entstehen können. Der MDR1-Genotyp eines Hundes ergibt sich aus der Kombination eines von väterlicher (+ oder -) und eines von mütterlicher Seite (+ oder -) vererbten Merkmals. „+“ steht dabei für ein intaktes MDR1-Gen und „-“ für ein defektes MDR1-Gen bezogen auf das Merkmal MDR1 nt230(del4). Für den MDR1-Genotyp eines Hundes gibt es drei verschiedene Möglichkeiten:
Nicht betroffen - MDR1(+/+), Merkmalsträger - MDR1(+/-) und Betroffen - MDR1(-/-).
Ist der MDR1-Genotyp zweier Zuchttiere bekannt, kann bereits eine theoretische Voraussage über die MDR1-Genotypen der Nachkommengeneration getroffen werden. Betroffene Tiere mit dem Genotyp MDR1(-/-) können aus einer Kreuzung der Genotypen MDR1(+/-) x MDR1(+/-), MDR1(+/-) x MDR1(-/-) oder MDR1(-/-) x MDR1(-/-) entstehen. Bei Kreuzung der Genotypen MDR1(+/+) x MDR1(-/-), MDR1(+/+) x MDR1(+/-), und MDR1(+/+) x MDR1(+/+) entstehen dagegen keine betroffenen MDR1(-/-) Tiere, aber außer bei MDR1(+/+) x MDR1(+/+) unter Umständen wieder Merkmalsträger.
Nachdem einige dem Verband für das Deutsche Hundewesen (VDH) angeschlossenen Zuchtvereine schon von sich aus diese Verpaarungsregeln beachtet hatten, hat der VDH diese den ihm angeschlossenen Collie- und Sheltiezuchtvereinen im Juni 2009 auferlegt. Ab November 2009 müssen nun sämtliche Zuchttiere auf den MDR1-Defekt getestet werden.
Ziel ist, keine vom Defekt betroffenen Nachkommen mehr zu erzeugen.
Dadurch kommt es zu einer mangelhaften oder fehlenden Synthese eines bestimmten Proteins (P-Glykoprotein, P-gp), welches ein wichtiger Bestandteil der Blut-Hirn-Schranke ist, was zu einer Überempfindlichkeit gegenüber manchen Arzneimitteln führt. Urheber dieses Defektes ist wahrscheinlich ein einziger Hund, der etwa Mitte des 19. Jahrhunderts gelebt hat und maßgeblich an der Entstehung und Festigung der Rasse Collie beteiligt war. Daher lässt sich dieser Defekt bei Hunderassen finden, die nachweisbar mit dem Collie verwandt sind.
Bei anderen vom Defekt betroffenen Rassen dient diese Mutation dann als Nachweis der Verwandtschaft. Ein funktionierendes MDR1-System ist vor allem bei Säugetieren (und Menschen) bekannt und hier evolutionsgeschichtlich sehr alt. Tiere, die dieses System nicht besitzen, können ähnliche Empfindlichkeiten für Medikamente zeigen.
Betroffene Rassen
Die Projektgruppe MDR1-Defekt beim Collie an der Justus-Liebig-Universität Gießen hat im Jahr 2004 im Rahmen einer Studie zur Häufigkeit des MDR1-Defektes bei verschiedenen Hunderassen Hunde aus 30 verschiedenen Rassen und 10 Europäischen Ländern getestet.
Der Defekt im MDR1-Gen wurde u. a. bei folgenden Hunderassen gefunden: Collie (Kurzhaarcollie und Langhaarcollie), Shetland Sheepdog, Australian Shepherd, Bobtail und Border-Collie. Obwohl bisher noch keine nt230(del4) Mutation beim Bearded Collie nachgewiesen werden konnte, lässt die geringe Probenzahl noch keine endgültige Bewertung zu. Bei den Rassen Wäller und Bobtail konnten bisher nur heterozygot von diesem Defekt betroffene Hunde (MDR1+/-) nachgewiesen werden, mit dem Auftreten homozygoter Mutationen müsste allerdings auch bei diesen Rassen gerechnet werden.
Beim Wäller ist es jedoch so, dass die Zuchttiere getestet sein müssen und bei der Verpaarung darauf geachtet wird, ein Trägertier nur mit einem freien zu verpaaren, damit es eben nicht zu homozygoten Welpen kommt.
Des Weiteren ist der Defekt bei folgenden Rassen bekannt: Deutscher Schäferhund, English Shepherd, McNab, Silken Windhound, Langhaarwhippet, Berger Blanc Suisse sowie einigen seltenen Rassen.
Seit Anfang 2008 werden außerdem weitere Rassen wie Barsoi, Belgischer Schäferhund oder Kelpie auf den Defekt untersucht.
Symptome, Diagnostik und Folgen für das Tier
Der Defekt im MDR1-Gen führt zu einer mangelhaften oder fehlenden Synthese des P-Glycoproteins. Dieses Eiweiß spielt eine Rolle bei ATP-abhängigen Transportvorgängen zwischen Blut und Gewebe und ist im Gehirn, in Leber, Nieren, Darm, Plazenta und Hoden zu finden. Neben seiner Funktion beim Transport körperfremder Stoffe, limitiert es den Transport der Hormone der Nebennierenrinde (Cortisol, Corticosteron) in das Gehirn und hat damit Einfluss auf die Hypothalamus-Hypophyse-Nebennierenrinden-Achse. Bei einem MDR1-Defekt kommt es daher zu einem erhöhten Übergang der Nebennierenrindenhormone in die übergeordneten Zentren und aufgrund des negativen Feedbacks zu erniedrigten Kortisolwerten im Blut.
Als eine weitere Folge des Defektes wird auch eine höhere Anfälligkeit für chronisch entzündliche Darmerkrankungen vermutet.
Bei nicht vom Defekt betroffenen Tieren dient dieses Protein u. a. dazu, körperfremde Stoffe wie Arzneimittel aus dem Körper herauszutransportieren. Es besteht also eine Art Resistenz gegenüber unerwünschten Nebenwirkungen – die sogenannte Multiple Drug Resistance.
Bekannt sind bisher die Auswirkungen auf die Blut-Hirn-Schranke. Bei dieser Grenze zwischen den Hirnblutgefässen und dem Hirnnervengewebe stellt ein sogenannter MDR1-Transporter eine Schutzbarriere für das Gehirn dar. Dieser Transporter ist Teil der Blut-Hirn-Schranke und befindet sich normalerweise auf der Oberfläche der Endothelzellen (Zellen, die die Wände der Blutgefäße auskleiden). Er sorgt dafür, dass toxische Verbindungen und Arzneistoffe in den Gehirnkapillaren zurückgehalten werden und nicht in das Gehirn eindringen können.
Besteht nun bei einem Hund der MDR1-Defekt, fehlt der Transporter und der Schutz funktioniert nicht mehr. Bei betroffenen Tieren können daher nach der Verabreichung von bestimmten Antiparasitika, Zytostatika, Durchfallmitteln oder Antibiotika starke neurotoxische Nebenwirkungen auftreten − bis zum Tod. Bei Mäusen, bei welchen der MDR1-Transporter bewusst ausgeschaltet wurde, traten nicht nur Ivermectin, sondern auch zahlreiche andere Arzneistoffe bis zu 90-fach mehr ins Gehirn als bei Vergleichstieren mit intakter Blut-Hirn-Schranke. Diese Stoffe sind auch eine potentielle Gefahr für einen vom MDR1-Defekt betroffenen Hund.
Mögliche Folge dieses Gendefektes ist die Überempfindlichkeit des Hundes gegenüber bestimmten Arzneistoffen. Da die Gabe einiger dieser Mittel bei betroffenen Hunden allerdings zum Tode führen kann, wird ein Gentest aller Hunde betroffener Rassen empfohlen. Die Universität Gießen bietet einen Test auf den MDR1-Defekt an. Zu diesem Zweck nimmt der Tierarzt dem zu untersuchenden Hund eine kleine Menge Blut ab (1 ml EDTA-Blut) und sendet diese Probe an die Universität. Mit Hilfe eines genetischen Tests wird die Blutprobe dann auf ein Vorliegen der MDR1-Mutation untersucht, dass Ergebnis wird dem Hundehalter mitgeteilt.
Ist der Hund vom Defekt betroffen, hat der Hundehalter einige Dinge zu beachten. So dürfen beispielsweise bestimmte Wurmkuren und Flohschutzmittel nicht mehr verabreicht werden. Auch bei Durchfall oder Herzerkrankungen eingesetzte Medikamente können weitreichende unerwünschte Nebenwirkungen haben. Bekannt ist eine Überempfindlichkeit z. B. für die Wirkstoffe Ivermectin, Doramectin, Moxidectin (nur bei oraler Anwendung) und Loperamid. Milbemycinoxim und Emodepsid dürfen nur unter exakter Dosierung eingesetzt werden. Viele weitere Wirkstoffe stehen aber in dem Verdacht, unerwünschte Nebenwirkungen hervorrufen zu können. Generell sollte daher der behandelnde Tierarzt über den Defekt informiert werden. Der Hund selber gilt als Risikopatient.
Bei Spaziergängen ist dann darauf zu achten, dass der Hund keinen Kot von beispielsweise Pferden zu sich nimmt, da dieser einen der gefährlichen Wirkstoffe in unveränderter Form enthalten kann.
Bisher sind Überempfindlichkeiten vor allen bei Hunden mit homozygoter Vererbung des MDR1-Defektes (MDR1 -/-) bekannt. Inzwischen wurden aber auch Reaktionen bei Trägern (MDR1 +/-) beobachtet. In einer US-amerikanischen Studie (siehe unter Links) werden die Träger deshalb als „sensitive“ bezeichnet, die vom Defekt betroffenen Hunde (MDR1 -/-) als „super sensitive“. Ebenfalls in den USA wurden die vom Defekt betroffenen Hunde inzwischen von der Forschung als Versuchstiere entdeckt.
Auswirkungen auf die Zucht
Aufgrund der Probleme in der Arzneitherapie von Hunden mit dem Genotyp MDR1(-/-) wird z. B. von der Universität in Gießen empfohlen, den Gendefekt in der Zucht betroffener Hunderassen zu berücksichtigen und so zu verpaaren, dass keine vom Defekt betroffenen Nachkommen entstehen können. Der MDR1-Genotyp eines Hundes ergibt sich aus der Kombination eines von väterlicher (+ oder -) und eines von mütterlicher Seite (+ oder -) vererbten Merkmals. „+“ steht dabei für ein intaktes MDR1-Gen und „-“ für ein defektes MDR1-Gen bezogen auf das Merkmal MDR1 nt230(del4). Für den MDR1-Genotyp eines Hundes gibt es drei verschiedene Möglichkeiten:
Nicht betroffen - MDR1(+/+), Merkmalsträger - MDR1(+/-) und Betroffen - MDR1(-/-).
Ist der MDR1-Genotyp zweier Zuchttiere bekannt, kann bereits eine theoretische Voraussage über die MDR1-Genotypen der Nachkommengeneration getroffen werden. Betroffene Tiere mit dem Genotyp MDR1(-/-) können aus einer Kreuzung der Genotypen MDR1(+/-) x MDR1(+/-), MDR1(+/-) x MDR1(-/-) oder MDR1(-/-) x MDR1(-/-) entstehen. Bei Kreuzung der Genotypen MDR1(+/+) x MDR1(-/-), MDR1(+/+) x MDR1(+/-), und MDR1(+/+) x MDR1(+/+) entstehen dagegen keine betroffenen MDR1(-/-) Tiere, aber außer bei MDR1(+/+) x MDR1(+/+) unter Umständen wieder Merkmalsträger.
Nachdem einige dem Verband für das Deutsche Hundewesen (VDH) angeschlossenen Zuchtvereine schon von sich aus diese Verpaarungsregeln beachtet hatten, hat der VDH diese den ihm angeschlossenen Collie- und Sheltiezuchtvereinen im Juni 2009 auferlegt. Ab November 2009 müssen nun sämtliche Zuchttiere auf den MDR1-Defekt getestet werden.
Ziel ist, keine vom Defekt betroffenen Nachkommen mehr zu erzeugen.
eingestellt von: ise; Quellennachweis
Megaösophagus

Der Megaösophagus (Speiseröhre) ist eine Erkrankung der Speiseröhre, die bei verschiedenen Haustierarten vorkommt und entweder erblich bedingt oder als Folge einer zugrundeliegenden Erkrankung auftreten kann. Ein Megaösophagus kann angeboren sein, kann aber prinzipiell auch in jedem Alter neu auftreten. Am häufigsten betroffen ist der Hund. In der Humanpathologie wird der Begriff in Zusammenhang mit der Infektion mit Trypanosoma cruzi bei der Chagas-Krankheit gebraucht.
Pathophysiologie
Ein angeborener Megaösophagus beruht auf einer mechanischen oder funktionellen Störung in der Durchgängigkeit der Speiseröhre, entweder als Folge einer bestehenden Striktur oder eines neuromuskulären Problems.
Dies führt dazu, dass die aufgenommene Nahrung nicht wie üblich durch die normale Peristaltik in den Magen gelangt, sondern in der Speiseröhre verbleibt. Durch mangelnden Muskeltonus und/oder den Druck des Inhalts der Speiseröhre auf die Speiseröhrenwand kommt es mit der Zeit zu einer Ausweitung (Dilatation) des Organs, was wiederum die Peristaltik und damit die Funktionalität immer weiter einschränkt (Teufelskreis).
Persistierender rechter Aortenbogen
Der persistierende rechte Aortenbogenist ein Sonderfall der angeborenen Striktur, welcher symptomatisch der Arteria lusoria beim Menschen ähnelt.
In der Embryonalentwicklung entwickelt sich die Aorta nicht wie üblich aus der linken, sondern aus der rechten vierten Kiemenbogenarterie. Das führt dazu, dass die Speiseröhre zwischen Aorta, Truncus pulmonalis und Ligamentum arteriosum eingeklemmt wird, wodurch es zu einer mechanischen Verengung der Speiseröhre kommt, welche wiederum mechanisch zu einer Dilatation führt, sobald die betroffenen Welpen entwöhnt werden und das Festfutter die Engstelle nicht passieren kann.
Erworbener Megaösophagus
Ein erworbener Megaösophagus kann als Folge einer Raumforderung eintreten (z. B. durch Tumoren oder Abszesse), welche auf die Speiseröhre drückt, durch eine erworbene Striktur beispielsweise als Folge einer Vernarbung entstehen oder auch eine Folge einer erworbenen neurologischen oder muskulären Störung (Polymyositis, Polymyopathie) sein.
Beim Hund entsteht er am häufigsten idiopathisch, also ohne erkennbare Ursache, aber auch eine erworbene Myasthenia gravis, ein systemischer Lupus erythematodes, eine Hiatushernie, eine Bleivergiftung, eine Speiseröhrenentzündung, eine Schilddrüsenunterfunktion, ein Thymom oder ein Nebennierenrindenunterfunktion können einen Megaösophagus verursachen.
Signalement
Welpen mit primärem angeborenem Megaösophagus werden meist im Alter von einigen Wochen bis einigen Monaten vorgestellt, teilweise aufgrund von anhaltendem Regurgitieren (vom Besitzer häufig als Erbrechen gedeutet) und Gewichtsverlust, teilweise auch aufgrund einer akuten Aspirationspneumonie. Überdurchschnittlich häufig von angeborenem Megaösophagus betroffene Rassen sind Shar-Pei, Foxterrier, Deutscher Schäferhund, Deutsche Dogge, Irish Setter, Labrador Retriever, Zwergschnauzer sowie Neufundländer.
Welpen mit persistierendem rechtem Aortenbogen entwickeln die Symptome bei der Umstellung von Milch auf feste Nahrung, die an der Engstelle stecken bleibt und die Speiseröhre mechanisch ausweitet. Überdurchschnittlich häufig von dieser Problematik betroffene Rassen sind Boston Terrier, Deutscher Schäferhund und Irish Setter.
Erworbener Megaösophagus kann in jedem Alter entweder als idiopathische Störung oder sekundär zu anderen Erkrankungen auftreten. Es existieren keine Prädispositionen nach Alter oder Rasse.
Symptome
Leitsymptom eines Megaösophagus ist die Regurgitation von Nahrung aus der Speiseröhre. Sie muss vom Erbrechen abgegrenzt werden, bei dem Nahrung aus dem Magen wieder ausgestoßen wird.
Dies geschieht normalerweise während der Anamnese, während der der Besitzer nach Zeichen von Erbrechen (Anzeichen von Übelkeit, Aufstoßen, Bauchpresse) gefragt wird. Die Regurgitation muss nicht unmittelbar nach der Nahrungsaufnahme erfolgen, sondern kann auch zeitlich verzögert auftreten.
Chronischer Megaösophagus führt zu Gewichtsverlust und Abmagerung. Als Komplikation kann es häufig auch zu einer Aspirationspneumonie kommen, wenn regurgitierte Nahrung in die Luftröhre gelangt und aspiriert wird.
Diagnose
Der Verdacht auf Megaösophagus ergibt sich aus der Anamnese und dem klinischen Bild.
Wird Druck auf den Bauch ausgeübt, kann sich die Speiseröhre im Hals sichtbar aufblähen. Eine Röntgenaufnahme des Brustkorbs zeigt eine geweitete, mit Luft und/oder Futter gefüllte Speiseröhre. Bei angeborenem oder erworbenem Megaösophagus ist die Weitung meist gleichmäßig; bei Megaösophagus als Folge eines persistierenden rechten Aortenbogens oder einer anderen Striktur ist die Speiseröhre nur vor der Engstelle aufgeweitet.
Therapie und Prognose
Eine kausale Behandlung ist beim persistierenden rechten Aortenbogen sowie beim sekundären erworbenen Megaösophaus möglich:
Im ersten Fall wird die mechanische Einengung der Speiseröhre durch eine chirurgische Durchtrennung des Ligamentum arteriosum behoben, im zweiten die Grunderkrankung (z. B. die Myasthenia gravis) behandelt. Bei einer erfolgreichen Behandlung einer Grunderkrankung ist die Prognose gut. Erfolgt ein chirurgischer Eingriff bei persistierendem rechtem Aortenbogen rechtzeitig, kann sich der Megaösophagus zurückbilden. Erfolgt der Eingriff zu spät, kann die Aufweitung trotz Beheben der Engstelle erhalten bleiben.
Ein angeborener Megaösophagus bildet sich gelegentlich vor dem Alter von 6 Monaten spontan zurück. Eine medikamentöse oder chirurgische Behandlung ist nicht möglich. Durch Ausprobieren kann der Besitzer diejenige Futterkonsistenz finden, die der betroffene Hund am besten verträgt. Die Fütterung sollte von erhöhter Stelle erfolgen und der Hund nach der Fütterung für 10-15 Minuten in derselben Position gehalten werden, damit die Schwerkraft die Futterpassage in den Magen unterstützen kann. Häufige kleine Mahlzeiten scheinen dabei am besten geeignet zu sein.
Die meisten Tiere mit einem angeborenen Megaösophagus, der sich nicht spontan zurückbildet, sterben schließlich an einer akuten Aspirationspneumonie oder an einer Lungenfibrose infolge von häufigen Aspirationspneumonien.
Genetik und Zuchthygiene
Der angeborene Megaösophagus ist erblich, wobei die Erbgänge sich je nach Rasse unterscheiden können. Beim Foxterrier ist der angeborene Megaösophagus als autosomal rezessiv beschrieben, beim Zwergschnauzer als autosomal rezessiv mit 60 % Penetranz.
Er kann auch als Teil einer angeborenen diffusen Erkrankung des Nervensystems auftreten, zum Beispiel beim Dalmatiner. Betroffene Tiere und ihre Eltern sind von der Zucht auszuschließen.
Der persistierende rechte Aortenbogen ist eine Hemmungsmissbildung der Kiemenbogenarterien, an der sowohl genetische Faktoren als auch Umwelteinflüsse beteiligt sind. Der Erbgang ist polygen. Betroffene Tiere sollten von der Zucht ausgeschlossen werden; eine Zuchtwertschätzung wäre wünschenswert.
Der erworbene Megaösophagus ist normalerweise nicht direkt erblich bedingt. Rassen mit einem erhöhten Risiko für Hypothyreose (Unterfunktion der Schilddrüse) und Addison-Syndrom haben allerdings durch diese Krankheiten bedingt ebenfalls ein erhöhtes Risiko für erworbenen Megaösophagus.
Pathophysiologie
Ein angeborener Megaösophagus beruht auf einer mechanischen oder funktionellen Störung in der Durchgängigkeit der Speiseröhre, entweder als Folge einer bestehenden Striktur oder eines neuromuskulären Problems.
Dies führt dazu, dass die aufgenommene Nahrung nicht wie üblich durch die normale Peristaltik in den Magen gelangt, sondern in der Speiseröhre verbleibt. Durch mangelnden Muskeltonus und/oder den Druck des Inhalts der Speiseröhre auf die Speiseröhrenwand kommt es mit der Zeit zu einer Ausweitung (Dilatation) des Organs, was wiederum die Peristaltik und damit die Funktionalität immer weiter einschränkt (Teufelskreis).
Persistierender rechter Aortenbogen
Der persistierende rechte Aortenbogenist ein Sonderfall der angeborenen Striktur, welcher symptomatisch der Arteria lusoria beim Menschen ähnelt.
In der Embryonalentwicklung entwickelt sich die Aorta nicht wie üblich aus der linken, sondern aus der rechten vierten Kiemenbogenarterie. Das führt dazu, dass die Speiseröhre zwischen Aorta, Truncus pulmonalis und Ligamentum arteriosum eingeklemmt wird, wodurch es zu einer mechanischen Verengung der Speiseröhre kommt, welche wiederum mechanisch zu einer Dilatation führt, sobald die betroffenen Welpen entwöhnt werden und das Festfutter die Engstelle nicht passieren kann.
Erworbener Megaösophagus
Ein erworbener Megaösophagus kann als Folge einer Raumforderung eintreten (z. B. durch Tumoren oder Abszesse), welche auf die Speiseröhre drückt, durch eine erworbene Striktur beispielsweise als Folge einer Vernarbung entstehen oder auch eine Folge einer erworbenen neurologischen oder muskulären Störung (Polymyositis, Polymyopathie) sein.
Beim Hund entsteht er am häufigsten idiopathisch, also ohne erkennbare Ursache, aber auch eine erworbene Myasthenia gravis, ein systemischer Lupus erythematodes, eine Hiatushernie, eine Bleivergiftung, eine Speiseröhrenentzündung, eine Schilddrüsenunterfunktion, ein Thymom oder ein Nebennierenrindenunterfunktion können einen Megaösophagus verursachen.
Signalement
Welpen mit primärem angeborenem Megaösophagus werden meist im Alter von einigen Wochen bis einigen Monaten vorgestellt, teilweise aufgrund von anhaltendem Regurgitieren (vom Besitzer häufig als Erbrechen gedeutet) und Gewichtsverlust, teilweise auch aufgrund einer akuten Aspirationspneumonie. Überdurchschnittlich häufig von angeborenem Megaösophagus betroffene Rassen sind Shar-Pei, Foxterrier, Deutscher Schäferhund, Deutsche Dogge, Irish Setter, Labrador Retriever, Zwergschnauzer sowie Neufundländer.
Welpen mit persistierendem rechtem Aortenbogen entwickeln die Symptome bei der Umstellung von Milch auf feste Nahrung, die an der Engstelle stecken bleibt und die Speiseröhre mechanisch ausweitet. Überdurchschnittlich häufig von dieser Problematik betroffene Rassen sind Boston Terrier, Deutscher Schäferhund und Irish Setter.
Erworbener Megaösophagus kann in jedem Alter entweder als idiopathische Störung oder sekundär zu anderen Erkrankungen auftreten. Es existieren keine Prädispositionen nach Alter oder Rasse.
Symptome
Leitsymptom eines Megaösophagus ist die Regurgitation von Nahrung aus der Speiseröhre. Sie muss vom Erbrechen abgegrenzt werden, bei dem Nahrung aus dem Magen wieder ausgestoßen wird.
Dies geschieht normalerweise während der Anamnese, während der der Besitzer nach Zeichen von Erbrechen (Anzeichen von Übelkeit, Aufstoßen, Bauchpresse) gefragt wird. Die Regurgitation muss nicht unmittelbar nach der Nahrungsaufnahme erfolgen, sondern kann auch zeitlich verzögert auftreten.
Chronischer Megaösophagus führt zu Gewichtsverlust und Abmagerung. Als Komplikation kann es häufig auch zu einer Aspirationspneumonie kommen, wenn regurgitierte Nahrung in die Luftröhre gelangt und aspiriert wird.
Diagnose
Der Verdacht auf Megaösophagus ergibt sich aus der Anamnese und dem klinischen Bild.
Wird Druck auf den Bauch ausgeübt, kann sich die Speiseröhre im Hals sichtbar aufblähen. Eine Röntgenaufnahme des Brustkorbs zeigt eine geweitete, mit Luft und/oder Futter gefüllte Speiseröhre. Bei angeborenem oder erworbenem Megaösophagus ist die Weitung meist gleichmäßig; bei Megaösophagus als Folge eines persistierenden rechten Aortenbogens oder einer anderen Striktur ist die Speiseröhre nur vor der Engstelle aufgeweitet.
Therapie und Prognose
Eine kausale Behandlung ist beim persistierenden rechten Aortenbogen sowie beim sekundären erworbenen Megaösophaus möglich:
Im ersten Fall wird die mechanische Einengung der Speiseröhre durch eine chirurgische Durchtrennung des Ligamentum arteriosum behoben, im zweiten die Grunderkrankung (z. B. die Myasthenia gravis) behandelt. Bei einer erfolgreichen Behandlung einer Grunderkrankung ist die Prognose gut. Erfolgt ein chirurgischer Eingriff bei persistierendem rechtem Aortenbogen rechtzeitig, kann sich der Megaösophagus zurückbilden. Erfolgt der Eingriff zu spät, kann die Aufweitung trotz Beheben der Engstelle erhalten bleiben.
Ein angeborener Megaösophagus bildet sich gelegentlich vor dem Alter von 6 Monaten spontan zurück. Eine medikamentöse oder chirurgische Behandlung ist nicht möglich. Durch Ausprobieren kann der Besitzer diejenige Futterkonsistenz finden, die der betroffene Hund am besten verträgt. Die Fütterung sollte von erhöhter Stelle erfolgen und der Hund nach der Fütterung für 10-15 Minuten in derselben Position gehalten werden, damit die Schwerkraft die Futterpassage in den Magen unterstützen kann. Häufige kleine Mahlzeiten scheinen dabei am besten geeignet zu sein.
Die meisten Tiere mit einem angeborenen Megaösophagus, der sich nicht spontan zurückbildet, sterben schließlich an einer akuten Aspirationspneumonie oder an einer Lungenfibrose infolge von häufigen Aspirationspneumonien.
Genetik und Zuchthygiene
Der angeborene Megaösophagus ist erblich, wobei die Erbgänge sich je nach Rasse unterscheiden können. Beim Foxterrier ist der angeborene Megaösophagus als autosomal rezessiv beschrieben, beim Zwergschnauzer als autosomal rezessiv mit 60 % Penetranz.
Er kann auch als Teil einer angeborenen diffusen Erkrankung des Nervensystems auftreten, zum Beispiel beim Dalmatiner. Betroffene Tiere und ihre Eltern sind von der Zucht auszuschließen.
Der persistierende rechte Aortenbogen ist eine Hemmungsmissbildung der Kiemenbogenarterien, an der sowohl genetische Faktoren als auch Umwelteinflüsse beteiligt sind. Der Erbgang ist polygen. Betroffene Tiere sollten von der Zucht ausgeschlossen werden; eine Zuchtwertschätzung wäre wünschenswert.
Der erworbene Megaösophagus ist normalerweise nicht direkt erblich bedingt. Rassen mit einem erhöhten Risiko für Hypothyreose (Unterfunktion der Schilddrüse) und Addison-Syndrom haben allerdings durch diese Krankheiten bedingt ebenfalls ein erhöhtes Risiko für erworbenen Megaösophagus.
eingestellt von: ise; Quellen- und Bildnachweis
x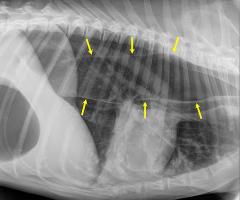
Quellen- und Bildnachweis
Bildname:
Megaösophagus - Erkrankung der Speiseröhre
Urheber:
Wikipedia/Anka Friedrich
Link:
Merle-Faktor

Der so genannte Merle-Faktor (kurz Merle) ist eine der vielen verschiedenen Farbvariationen des Fells bei Hunden und besonders in der Colliezucht stark verbreitet. Die Grundfarbe des Felles ist bei Merles stellenweise aufgehellt in der Weise, dass unregelmäßige, zerrissen wirkende Flecken in der Grundfarbe auf einem aufgehellten Grund zu sehen sind.
Rassen
Merle kommt bei Deutschen Doggen und Dackeln der Farbe „Tiger“, Collies, Shelties und anderen mit der Farb-Bezeichnung Blue Merle, Corgies, Foxhoundschläge, Catahoula Leopard Dog, Dunkerhunden, einigen Hütehundeschlägen wie Bergamasker Hirtenhund, Border-Collies, Bobtails, Australian Shepherds und Farbschlägen des Altdeutschen Hütehundes, dem Beauceron und dem Chihuahua vor.
Genetik
Das Merle ist auf ein Gen (Merle-Gen) im Erbgut des Hundes zurückzuführen. Es ist eine Mutation des Silver-Locus-Gens (Pmel17), das sich beim Haushund auf Chromosom CFA10 befindet.
Das Merle-Gen hellt nur Eumelanin auf, während es Fellbereiche, in denen ausschließlich Phäomelanin vorkommt, unverändert lässt.
Wenn man einen heterozygoten Träger des Merle-Gens mit einem Hund paart, der das Gen nicht hat, sind statistisch 50 % Merle- und 50 % andersfarbige Welpen zu erwarten. In der Praxis fallen normalerweise unter 50 % Merle-Welpen.
Aussehen
Eumelanin verursacht die schwarze Fellfarbe. Durch eine Mutation des Braun-Locus entsteht ein unvollständiger okulokutaner Albinismus Typ 3 (OCA3). Dadurch werden Fellbereiche, in denen ausschließlich Eumelanin produziert wird, zu Braun aufgehellt.
Die auf Eumelanin beruhenden Farben wie Schwarz und Braun können durch das Merle-Gen aufgehellt werden. In den so gefärbten Bereichen oder einzelnen schwarzen oder braunen Haaren entstehen weiße Abzeichen. Sie können als große Flecken oder als feine Sprenkelung vorkommen. Manchmal wird das Schwarz auch zu einem Grau aufgehellt, das von einem dunklen Graublau über rosastichig bis hin zu einem zarten Hellgrau variieren kann. Entsprechend kann das durch den Braun-Locus entstandene Braun zu helleren Brauntönen aufgehellt werden.
Die Augen können einfarbig braun, einfarbig blau, oder gemischtfarbig sein – wobei es Hunde gibt, die ein blaues und ein braunes Auge haben, als auch solche, die beide Farben in einem Auge vereinen.
Das „Allel e“ (englisch recessive yellow) des Extension-Locus bewirkt, dass ein Hund am gesamten Körper nur das gelb oder braun aussehende Phäomelanin produziert und deshalb einfarbig rotbraun bis goldfarben ist. Da das Merle-Gen Phäomelanin nicht beeinflussen kann, zeigen Hunde mit dieser braunen Farbe an keiner Stelle des Körpers die typische Merle-Zeichnung. In Rassen wo Merle vorkommt, ist deshalb bei Tieren dieser Farbe Vorsicht geboten, da man sonst unwissentlich zwei Träger der Merle-Gens miteinander verpaaren könnte. Durch Phäomelanin braun gefärbte Fellbereiche bei anderen Fellzeichnungen, wie der Brand bei Hunden mit roter Zeichnung in Gesicht und an Beinen oder wie die hellen Haare im Fell der geflammten Hunde werden ebenfalls nicht durch das Merle-Gen aufgehellt.
Gesundheitliche Auswirkungen der homozygoten Form
Das Merle-Gen wird intermediär vererbt. Hunde, die das Gen nur einmal aufweisen, also heterozygot sind, sind meist gesund. Das Merle-Gen führt neben einem größeren Weißanteil im Fell in einigen Fällen zu Fehlbildungen des Innenohrs mit Taubheit.
Bei reinerbigen (homozygoten) Tieren sind 10 % einseitig und 15 % auf beiden Ohren taub. Von den mischerbigen Tieren sind 2,7 % einseitig, 0,9 % vollständig taub. Vor allem bei homozygoten Tieren können auch Fehlbildungen der Augen auftreten. Betroffene Tiere können in der Entwicklung hinter Wurfgeschwistern zurückbleiben, verminderte Lebensfreude zeigen und sterben manchmal vor der Geschlechtsreife.
Aus Tierschutzgründen ist daher die Verpaarung zweier Träger des Merle-Faktors nicht zu empfehlen. Die gezielte Zucht mit einem Gendefekt aus rein ästhetischen Beweggründen ist stark umstritten. Im „Gutachten zur Auslegung von §11 des Tierschutzgesetzes (Verbot von Qualzuchten)“ (BMELV) wird generell die Empfehlung ausgesprochen, auf die Zucht mit dem Merle-Gen zu verzichten.
Rassen
Merle kommt bei Deutschen Doggen und Dackeln der Farbe „Tiger“, Collies, Shelties und anderen mit der Farb-Bezeichnung Blue Merle, Corgies, Foxhoundschläge, Catahoula Leopard Dog, Dunkerhunden, einigen Hütehundeschlägen wie Bergamasker Hirtenhund, Border-Collies, Bobtails, Australian Shepherds und Farbschlägen des Altdeutschen Hütehundes, dem Beauceron und dem Chihuahua vor.
Genetik
Das Merle ist auf ein Gen (Merle-Gen) im Erbgut des Hundes zurückzuführen. Es ist eine Mutation des Silver-Locus-Gens (Pmel17), das sich beim Haushund auf Chromosom CFA10 befindet.
Das Merle-Gen hellt nur Eumelanin auf, während es Fellbereiche, in denen ausschließlich Phäomelanin vorkommt, unverändert lässt.
Wenn man einen heterozygoten Träger des Merle-Gens mit einem Hund paart, der das Gen nicht hat, sind statistisch 50 % Merle- und 50 % andersfarbige Welpen zu erwarten. In der Praxis fallen normalerweise unter 50 % Merle-Welpen.
Aussehen
Eumelanin verursacht die schwarze Fellfarbe. Durch eine Mutation des Braun-Locus entsteht ein unvollständiger okulokutaner Albinismus Typ 3 (OCA3). Dadurch werden Fellbereiche, in denen ausschließlich Eumelanin produziert wird, zu Braun aufgehellt.
Die auf Eumelanin beruhenden Farben wie Schwarz und Braun können durch das Merle-Gen aufgehellt werden. In den so gefärbten Bereichen oder einzelnen schwarzen oder braunen Haaren entstehen weiße Abzeichen. Sie können als große Flecken oder als feine Sprenkelung vorkommen. Manchmal wird das Schwarz auch zu einem Grau aufgehellt, das von einem dunklen Graublau über rosastichig bis hin zu einem zarten Hellgrau variieren kann. Entsprechend kann das durch den Braun-Locus entstandene Braun zu helleren Brauntönen aufgehellt werden.
Die Augen können einfarbig braun, einfarbig blau, oder gemischtfarbig sein – wobei es Hunde gibt, die ein blaues und ein braunes Auge haben, als auch solche, die beide Farben in einem Auge vereinen.
Das „Allel e“ (englisch recessive yellow) des Extension-Locus bewirkt, dass ein Hund am gesamten Körper nur das gelb oder braun aussehende Phäomelanin produziert und deshalb einfarbig rotbraun bis goldfarben ist. Da das Merle-Gen Phäomelanin nicht beeinflussen kann, zeigen Hunde mit dieser braunen Farbe an keiner Stelle des Körpers die typische Merle-Zeichnung. In Rassen wo Merle vorkommt, ist deshalb bei Tieren dieser Farbe Vorsicht geboten, da man sonst unwissentlich zwei Träger der Merle-Gens miteinander verpaaren könnte. Durch Phäomelanin braun gefärbte Fellbereiche bei anderen Fellzeichnungen, wie der Brand bei Hunden mit roter Zeichnung in Gesicht und an Beinen oder wie die hellen Haare im Fell der geflammten Hunde werden ebenfalls nicht durch das Merle-Gen aufgehellt.
Gesundheitliche Auswirkungen der homozygoten Form
Das Merle-Gen wird intermediär vererbt. Hunde, die das Gen nur einmal aufweisen, also heterozygot sind, sind meist gesund. Das Merle-Gen führt neben einem größeren Weißanteil im Fell in einigen Fällen zu Fehlbildungen des Innenohrs mit Taubheit.
Bei reinerbigen (homozygoten) Tieren sind 10 % einseitig und 15 % auf beiden Ohren taub. Von den mischerbigen Tieren sind 2,7 % einseitig, 0,9 % vollständig taub. Vor allem bei homozygoten Tieren können auch Fehlbildungen der Augen auftreten. Betroffene Tiere können in der Entwicklung hinter Wurfgeschwistern zurückbleiben, verminderte Lebensfreude zeigen und sterben manchmal vor der Geschlechtsreife.
Aus Tierschutzgründen ist daher die Verpaarung zweier Träger des Merle-Faktors nicht zu empfehlen. Die gezielte Zucht mit einem Gendefekt aus rein ästhetischen Beweggründen ist stark umstritten. Im „Gutachten zur Auslegung von §11 des Tierschutzgesetzes (Verbot von Qualzuchten)“ (BMELV) wird generell die Empfehlung ausgesprochen, auf die Zucht mit dem Merle-Gen zu verzichten.
eingestellt von: ise; Quellen- und Bildnachweis
x
Quellen- und Bildnachweis
Bildname:
Dooggs | Australian Shepherd
Urheber:
Wikipedia/Bonnie van den Born
Link:
Myasthenia gravis

Die Myasthenia gravis pseudoparalytica (von „Schwäche“, „schwer“,„falsch“ und „Lähmung“; Kürzel: MG) bzw. laut ICD-10 vereinfacht Myasthenia gravis, gehört zu einer Gruppe von neurologischen Erkrankungen, die durch eine gestörte Signalübertragung zwischen Nerv und Muskel gekennzeichnet sind, und als Störungen der neuromuskulären Erregungsübertragung oder als myasthene Syndrome zusammengefasst werden.
Sie ist eine Autoimmunerkrankung, bei der eine Störung an der motorischen Endplatte der quergestreiften Muskulatur (Skelettmuskulatur) vorliegt, deren Ursache aber nicht völlig erforscht ist.
Das Krankheitsbild der Myasthenia gravis ist gekennzeichnet durch eine belastungsabhängige Muskelschwäche die die oben genannte Muskulatur häufig asymmetrisch befällt, auch einzelne oder mehrere Muskeln, unabhängig von der Körperhälfte. Zur Hauptsymptomatik gehört ihre wechselnde Ausprägung, beispielsweise neben dem Wechsel betroffener Muskeln, kann sie oft im Tagesverlauf spontan ohne erkennbaren Grund zunehmen und/oder es kann eine Erholung der betroffenen Muskeln in Ruhe, ebenso plötzlich wieder eintreten.
Die Myasthenia gravis kommt sowohl bei Menschen wie auch bei Tieren, besonders bei Haushunden vor.
Sie ist eine Autoimmunerkrankung, bei der eine Störung an der motorischen Endplatte der quergestreiften Muskulatur (Skelettmuskulatur) vorliegt, deren Ursache aber nicht völlig erforscht ist.
Das Krankheitsbild der Myasthenia gravis ist gekennzeichnet durch eine belastungsabhängige Muskelschwäche die die oben genannte Muskulatur häufig asymmetrisch befällt, auch einzelne oder mehrere Muskeln, unabhängig von der Körperhälfte. Zur Hauptsymptomatik gehört ihre wechselnde Ausprägung, beispielsweise neben dem Wechsel betroffener Muskeln, kann sie oft im Tagesverlauf spontan ohne erkennbaren Grund zunehmen und/oder es kann eine Erholung der betroffenen Muskeln in Ruhe, ebenso plötzlich wieder eintreten.
Die Myasthenia gravis kommt sowohl bei Menschen wie auch bei Tieren, besonders bei Haushunden vor.
eingestellt von: ise; Quellen- und Bildnachweis
Nekrotisierende Myelopathie des Kooiker-Hundes
Die Nekrotisierende Myelopathie des Kooiker-Hundes ist eine innerhalb der ersten drei Lebensmonate bei Kooiker-Hunden auftretende Degeneration der weißen Substanz, vor allem im Halsmark. Sie äußert sich in gesteigerten Reflexen und schnell fortschreitenden Lähmungen.
eingestellt von: ise; Quellennachweis
Osteochondrosis
Eine Osteochondrose oder Osteochondrosis ist eine Störung der chondralen Ossifikation, also der Umwandlung von Knorpel zu Knochen als Teil des normalen Wachstumsprozesses.
Sie kann daher grundsätzlich an zwei Lokalisationen auftreten: im Gelenk und in den Wachstumsfugen.
In beiden Fällen wird der Knorpel nicht genügend rasch zu Knochen umgebaut, so dass sich eine abnorm dicke Knorpelschicht bilden kann. Da Knorpel keine eigenen Blutgefäße besitzt, sondern durch Diffusion ernährt wird, werden die tiefer gelegenen Knorpelschichten immer schlechter ernährt und degenerieren. Dabei kann sich bei der gelenksansässigen Osteochondrose auch ein Knorpelstück lösen und frei im Gelenk schwimmen („Gelenkmaus“) – man spricht dann von einer Osteochondrosis (oder Osteochondritis) dissecans, kurz OCD.
Osteochondrosen treten auch in der Veterinärmedizin (insbesondere bei großen Hunderassen) auf. Sie folgen dort meist einem polygenen Erbgang mit Umweltinteraktion bei einer Heritabilität zwischen 0,25 und 0,45.
Die Osteochondrose der Wirbelsäule wird nach Modic I-III eingeteilt. Spezialformen der Osteochondrose sind zum Beispiel:
- Osteochondrosis dissecans
- Osteochondrosis intervertebralis
- Morbus Scheuermann
- Morbus Perthes – aseptische Femurkopfnekrose
- Ellbogendysplasie bei Hunden
- Short Radius/Short Ulna Syndrom
- Osteochondrosis pubica
Sie kann daher grundsätzlich an zwei Lokalisationen auftreten: im Gelenk und in den Wachstumsfugen.
In beiden Fällen wird der Knorpel nicht genügend rasch zu Knochen umgebaut, so dass sich eine abnorm dicke Knorpelschicht bilden kann. Da Knorpel keine eigenen Blutgefäße besitzt, sondern durch Diffusion ernährt wird, werden die tiefer gelegenen Knorpelschichten immer schlechter ernährt und degenerieren. Dabei kann sich bei der gelenksansässigen Osteochondrose auch ein Knorpelstück lösen und frei im Gelenk schwimmen („Gelenkmaus“) – man spricht dann von einer Osteochondrosis (oder Osteochondritis) dissecans, kurz OCD.
Osteochondrosen treten auch in der Veterinärmedizin (insbesondere bei großen Hunderassen) auf. Sie folgen dort meist einem polygenen Erbgang mit Umweltinteraktion bei einer Heritabilität zwischen 0,25 und 0,45.
Die Osteochondrose der Wirbelsäule wird nach Modic I-III eingeteilt. Spezialformen der Osteochondrose sind zum Beispiel:
- Osteochondrosis dissecans
- Osteochondrosis intervertebralis
- Morbus Scheuermann
- Morbus Perthes – aseptische Femurkopfnekrose
- Ellbogendysplasie bei Hunden
- Short Radius/Short Ulna Syndrom
- Osteochondrosis pubica
eingestellt von: ise; Quellennachweis
Progressive Retinaatrophie
Bei der progressiven Retinaatrophie (PRA) handelt es sich um ein langsam fortschreitendes Absterben der Netzhaut von Hunden.
Es handelt sich hierbei um eine erbliche, progressive (stufenweise fortschreitende) und letztlich zur Erblindung beider Augen führende Erkrankung, wobei die degenerativen Prozesse im Bereich der Photorezeptoren beginnen und im Verlauf der Erkrankung die gesamte Netzhaut mit einbeziehen. Trotz des ähnlichen klinischen Erscheinungsbildes kann der Symptomatik eine Vielzahl verschiedener Ursachen zugrunde liegen. Die Progressive Retinaatrophie entspricht der Retinopathia pigmentosa des Menschen.
Verbreitung
Die Erkrankung betrifft verschiedenste Hunderassen und deren Mischlinge.
Sie wurde nachgewiesen bei Zwerg- und Mittelpudel, Amerikanischem Cocker, Englischem Cocker, Portugiesischem Wasserhund, Labrador Retriever, Samojede, English Setter, Rauhaar- und Langhaardackel, Tibet-Terrier, Tibet Spaniel, Akita Inu, Siberian Husky, Afghane, Australian Cattle Dog, Malinois, Bernhardiner, Border Collie, Cavalier King Charles Spaniel, Chesapeake Bay Retriever, Collie, English Springer Spaniel, Entlebucher Sennenhund, Golden Retriever, Irish Setter, Irish Terrier, Malteser, Mastiff, Nova Scotia Duck Tolling Retriever, Riesenschnauzer, Rottweiler, Saluki, Sloughi, Pyrenäenberghund, Bologneser, Neufundländer, PON, Saarlooswolfhund, Scottish Terrier, Schapendoes, Do Khyi, Sheltie, Zwergspitz sowie Kreuzungen zwischen Spaniels und Pudeln.
Ursache
Die Ursache liegt in den meisten Fällen in einem autosomal-rezessiv vererbten Gendefekt. Bei Mastiffs ist eine dominant vererbte Form beschrieben. Huskys und Samojeden bilden eine ans X-Chromosom gebundene Variante aus.
Behandlung
Bis heute gibt es keinerlei Medikamente oder operative Maßnahmen, um die PRA zu heilen. Hunde, die an der PRA erkranken und erblinden fast immer.
Genetischer Nachweis
Für eine Reihe von Hunderassen ist ein Gentest zum Nachweis der PRA verfügbar. Dazu gehören Irish Setter, Welsh Corgi, Sloughi, Bullterrier, Mastiff und Bullmastiff.
Es handelt sich hierbei um eine erbliche, progressive (stufenweise fortschreitende) und letztlich zur Erblindung beider Augen führende Erkrankung, wobei die degenerativen Prozesse im Bereich der Photorezeptoren beginnen und im Verlauf der Erkrankung die gesamte Netzhaut mit einbeziehen. Trotz des ähnlichen klinischen Erscheinungsbildes kann der Symptomatik eine Vielzahl verschiedener Ursachen zugrunde liegen. Die Progressive Retinaatrophie entspricht der Retinopathia pigmentosa des Menschen.
Verbreitung
Die Erkrankung betrifft verschiedenste Hunderassen und deren Mischlinge.
Sie wurde nachgewiesen bei Zwerg- und Mittelpudel, Amerikanischem Cocker, Englischem Cocker, Portugiesischem Wasserhund, Labrador Retriever, Samojede, English Setter, Rauhaar- und Langhaardackel, Tibet-Terrier, Tibet Spaniel, Akita Inu, Siberian Husky, Afghane, Australian Cattle Dog, Malinois, Bernhardiner, Border Collie, Cavalier King Charles Spaniel, Chesapeake Bay Retriever, Collie, English Springer Spaniel, Entlebucher Sennenhund, Golden Retriever, Irish Setter, Irish Terrier, Malteser, Mastiff, Nova Scotia Duck Tolling Retriever, Riesenschnauzer, Rottweiler, Saluki, Sloughi, Pyrenäenberghund, Bologneser, Neufundländer, PON, Saarlooswolfhund, Scottish Terrier, Schapendoes, Do Khyi, Sheltie, Zwergspitz sowie Kreuzungen zwischen Spaniels und Pudeln.
Ursache
Die Ursache liegt in den meisten Fällen in einem autosomal-rezessiv vererbten Gendefekt. Bei Mastiffs ist eine dominant vererbte Form beschrieben. Huskys und Samojeden bilden eine ans X-Chromosom gebundene Variante aus.
Behandlung
Bis heute gibt es keinerlei Medikamente oder operative Maßnahmen, um die PRA zu heilen. Hunde, die an der PRA erkranken und erblinden fast immer.
Genetischer Nachweis
Für eine Reihe von Hunderassen ist ein Gentest zum Nachweis der PRA verfügbar. Dazu gehören Irish Setter, Welsh Corgi, Sloughi, Bullterrier, Mastiff und Bullmastiff.
eingestellt von: ise; Quellennachweis
Sebadenitis

Die Sebadenitis (engl. sebaceous adenitis, SA; auch granulomatöse Sebadenitis, primäre Sebadenitis oder aseptische Sebadenitis) ist eine Erkrankung beim Hund, bei der die Talgdrüsen der Haut durch eine Entzündungsreaktion irreversibel zerstört werden.
Sie gehört zu den idiopathischen Erkrankungen, also zu den Krankheiten, deren eigentliche Ursache unbekannt ist. Es wird aber vermutet, dass die Sebadenitis genetisch bedingt ist. Sebadenitis ist abgeleitet von lateinisch Glandula sebacea, Talgdrüse, und Adenitis, Drüsenentzündung.
Pathophysiologie
Es gibt verschiedene Hypothesen zu den Ursachen der Erkrankung. Diese reichen von einer genetisch bedingten Entwicklungsstörung der Talgdrüsen über die Hypothese von Autoimmunreaktionen, die die Talgdrüsen zerstören zu Keratinisierungsstörungen, die zu einer Drüsenentzündung führen. Auch eine genetisch bedingte Immunerkrankung ist eine mögliche Erklärung.
Die Erkrankung geht zunächst mit einer Entzündung der Talgdrüsen einher. Später gehen diese zugrunde, was zu einer Hyperkeratose führt. Die Haarfollikel sind in ihrer Entwicklung beeinträchtigt, werden aber nicht völlig zerstört.
Klinik
Signalement
Die Erkrankung ist bei Hunden wie folgt beschrieben.
Bei Hunden tritt sie bei den Rassen Pudel, Magyar Vizsla und einigen nordischen Rassen wie Akita und Samojede häufiger auf, auch bei Hovawart, Schnauzer, Deutscher Schäferhund, Berner Sennenhund, Malteser, English Springer Spaniel und Mischlingen kommt sie vor. Die Erkrankung tritt bei einigen Rassen und Geschwistertieren gehäuft auf, beispielsweise beim Akita und Hovawart. Insgesamt wurden Fälle bei über 50 Rassen beschrieben.
Symptome
Die Erkrankung äußert sich durch fortschreitenden Haarausfall, häufig zunächst lokal begrenzt. Haarlose Stellen sind trocken und schuppig. Haarbüschel sind oft miteinander verklebt und haben Keratinmanschetten.
Typisch ist auch eine übermäßige Verhornung der Haut (Hyperkeratose). Bei verschiedenen Rassen gibt es unterschiedliche typische Krankheitsverläufe. Juckreiz ist nicht immer vorhanden und unterschiedlich stark ausgeprägt, er tritt erst bei Sekundärinfektionen auf.
Perakuten Erkrankungsverläufen gehen häufig Magen-Darm-Probleme voraus, gelegentlich kommen sie zusammen mit Futtermittelallergien vor. Viele betroffene Tiere haben Entzündungen des Gehörgangs durch vermehrte Bildung von Ohrenschmalz (Otitis externa ceruminosa).
Diagnose
Die Diagnosestellung erfolgt durch die Entnahme einer Hautbiopsie mit anschliessender Histologie und Nachweis der fehlenden Talgdrüsen und/oder der lymphozytären Entzündung in dem Bereich. Differentialdiagnostisch müssen vor allem Infektionen durch Bakterien, Parasiten und Pilze ausgeschlossen werden. Auch Malassezien-Dermatitis und Vitamin-A-responsive Dermatose kommen differentialdiagnostisch in Betracht.
Neben der aseptischen Form gibt es eine sekundäre Form der Sebadenitis, die durch Leishmaniose ausgelöst wird. Anders als bei der primären Form können sich hier die Talgdrüsen regenerieren. Einige Autoren grenzen diese Form jedoch von der Sebadenitis ab, zählen sie also genau nicht dazu.
Therapie und Prognose
Eine ursächliche Therapie existiert nicht. Die Gabe von Ciclosporin A kann die Erkrankung aufhalten, nach Absetzen schreitet die Zerstörung der Talgdrüsen jedoch weiter fort.
Bei milden Erkrankungsformen kann eine Therapie mit Shampoos, die keratinlösende Wirkstoffe enthalten, erfolgreich sein.
Es scheint, dass eine Behandlung in der (frühen) entzündlichen Phase der Erkrankung erfolgversprechender ist als in der (späten) chronischen Phase.
Es gibt Autoren, die aus diesem Grund empfehlen, eine Behandlung mit Ciclosporin so früh wie möglich zu beginnen. In einer internationalen Studie konnte kein Unterschied zwischen dem Effekt einer lokalen Behandlung und demjenigen einer systemischen Therapie nachgewiesen werden.
Genetik und Zuchthygiene
Der genaue Erbgang der Sebadenitis ist nicht bekannt; ihr Auftreten bei bestimmten Rassen und bei Geschwistern betroffener Hunde spricht aber dafür, dass es sich bei ihr um eine Erbkrankheit handelt.
Betroffene Tiere sollten deswegen nicht zur Zucht verwendet werden.
Sie gehört zu den idiopathischen Erkrankungen, also zu den Krankheiten, deren eigentliche Ursache unbekannt ist. Es wird aber vermutet, dass die Sebadenitis genetisch bedingt ist. Sebadenitis ist abgeleitet von lateinisch Glandula sebacea, Talgdrüse, und Adenitis, Drüsenentzündung.
Pathophysiologie
Es gibt verschiedene Hypothesen zu den Ursachen der Erkrankung. Diese reichen von einer genetisch bedingten Entwicklungsstörung der Talgdrüsen über die Hypothese von Autoimmunreaktionen, die die Talgdrüsen zerstören zu Keratinisierungsstörungen, die zu einer Drüsenentzündung führen. Auch eine genetisch bedingte Immunerkrankung ist eine mögliche Erklärung.
Die Erkrankung geht zunächst mit einer Entzündung der Talgdrüsen einher. Später gehen diese zugrunde, was zu einer Hyperkeratose führt. Die Haarfollikel sind in ihrer Entwicklung beeinträchtigt, werden aber nicht völlig zerstört.
Klinik
Signalement
Die Erkrankung ist bei Hunden wie folgt beschrieben.
Bei Hunden tritt sie bei den Rassen Pudel, Magyar Vizsla und einigen nordischen Rassen wie Akita und Samojede häufiger auf, auch bei Hovawart, Schnauzer, Deutscher Schäferhund, Berner Sennenhund, Malteser, English Springer Spaniel und Mischlingen kommt sie vor. Die Erkrankung tritt bei einigen Rassen und Geschwistertieren gehäuft auf, beispielsweise beim Akita und Hovawart. Insgesamt wurden Fälle bei über 50 Rassen beschrieben.
Symptome
Die Erkrankung äußert sich durch fortschreitenden Haarausfall, häufig zunächst lokal begrenzt. Haarlose Stellen sind trocken und schuppig. Haarbüschel sind oft miteinander verklebt und haben Keratinmanschetten.
Typisch ist auch eine übermäßige Verhornung der Haut (Hyperkeratose). Bei verschiedenen Rassen gibt es unterschiedliche typische Krankheitsverläufe. Juckreiz ist nicht immer vorhanden und unterschiedlich stark ausgeprägt, er tritt erst bei Sekundärinfektionen auf.
Perakuten Erkrankungsverläufen gehen häufig Magen-Darm-Probleme voraus, gelegentlich kommen sie zusammen mit Futtermittelallergien vor. Viele betroffene Tiere haben Entzündungen des Gehörgangs durch vermehrte Bildung von Ohrenschmalz (Otitis externa ceruminosa).
Diagnose
Die Diagnosestellung erfolgt durch die Entnahme einer Hautbiopsie mit anschliessender Histologie und Nachweis der fehlenden Talgdrüsen und/oder der lymphozytären Entzündung in dem Bereich. Differentialdiagnostisch müssen vor allem Infektionen durch Bakterien, Parasiten und Pilze ausgeschlossen werden. Auch Malassezien-Dermatitis und Vitamin-A-responsive Dermatose kommen differentialdiagnostisch in Betracht.
Neben der aseptischen Form gibt es eine sekundäre Form der Sebadenitis, die durch Leishmaniose ausgelöst wird. Anders als bei der primären Form können sich hier die Talgdrüsen regenerieren. Einige Autoren grenzen diese Form jedoch von der Sebadenitis ab, zählen sie also genau nicht dazu.
Therapie und Prognose
Eine ursächliche Therapie existiert nicht. Die Gabe von Ciclosporin A kann die Erkrankung aufhalten, nach Absetzen schreitet die Zerstörung der Talgdrüsen jedoch weiter fort.
Bei milden Erkrankungsformen kann eine Therapie mit Shampoos, die keratinlösende Wirkstoffe enthalten, erfolgreich sein.
Es scheint, dass eine Behandlung in der (frühen) entzündlichen Phase der Erkrankung erfolgversprechender ist als in der (späten) chronischen Phase.
Es gibt Autoren, die aus diesem Grund empfehlen, eine Behandlung mit Ciclosporin so früh wie möglich zu beginnen. In einer internationalen Studie konnte kein Unterschied zwischen dem Effekt einer lokalen Behandlung und demjenigen einer systemischen Therapie nachgewiesen werden.
Genetik und Zuchthygiene
Der genaue Erbgang der Sebadenitis ist nicht bekannt; ihr Auftreten bei bestimmten Rassen und bei Geschwistern betroffener Hunde spricht aber dafür, dass es sich bei ihr um eine Erbkrankheit handelt.
Betroffene Tiere sollten deswegen nicht zur Zucht verwendet werden.
eingestellt von: ise; Quellen- und Bildnachweis
Trachealkollaps des Hundes

Als Trachealkollaps bezeichnet man bei Haushunden ein Zusammenfallen der Luftröhre (lat. Trachea), durch das Erweichen der stützenden Knorpelspangen. Daraus resultiert eine Abnahme vor allem des Vertikaldurchmessers und somit eine Verengung der Luftröhre, die zu schweren Atemproblemen führen kann. Der Trachealkollaps der Hunde kann zumeist lange Zeit konservativ mit Medikamenten beherrscht werden, eine chirurgische Therapie ist möglich, allerdings aufwändig und nicht in jedem Fall erfolgreich.
Vorkommen und Ursachen
Ein Trachealkollaps kommt vor allem bei Haushunden, insbesondere bei zwergwüchsigen Hunderassen wie z.B. bei Yorkshireterrier, Chihuahua, Malteser, Zwergspitz und Zwergschnauzer vor. Vor allem Tiere mittleren Alters sind betroffen.
Die Ursache der Erkrankung ist bislang nicht geklärt. Vermutlich ist der Trachealkollaps genetisch bedingt (Erbkrankheit). Aber auch die Genese durch das Zusammentreffen verschiedener Faktoren (multifaktoriell) ist nicht ausgeschlossen. Die Erkrankung kann durch eine Reihe anderer Faktoren, darunter Infektionen der Atemwege, Allergien, Verengungen der Luftröhre, toxische Stäube und Dämpfe sowie Herzinsuffizienz, begünstigt werden.
Pathogenese
Die Knorpelerweichung kommt durch Veränderungen der Knorpelgrundsubstanz, vor allem durch einen verminderten Gehalt an Glykosaminoglykanen (insbesondere Chondroitinsulfat) und Glykoproteinen zustande, wodurch die Wasserbindungskapazität des Knorpels und damit dessen Elastizität sinkt. Die Zellzahl im Knorpel ist erniedrigt und die Grundsubstanz ist porös. Die Abnahme des Luftröhreninnendurchmessers geht mit einem erhöhten Strömungswiderstand der Luft einher und führt zu Turbulenzen.
Sekundär kommt es durch den Trachealkollaps zu einer Degeneration des Flimmerepithels, zu einer Hypertrophie der Drüsen in der Schleimhaut der Luftröhre, gelegentlich auch zur Bildung von Polypen.
Klinik
Das klinische Erscheinungsbild ist sehr variabel, ein Trachealkollaps kann lange symptomlos bleiben und der Grad der Verengung muss nicht mit dem Ausmaß klinischer Erscheinungen korrelieren. Die Symptome entwickeln sich allmählich und die Erkrankung schreitet langsam voran.
Eine typische Früherscheinung ist ein anfallsartiger Husten, der vor allem bei Aufregung oder stärkerer körperlicher Belastung auftritt. Auch ein Halsband kann bei starkem Zug an der Leine Druck auf die Luftröhre ausüben und somit die Hustenanfälle auslösen. Der Husten tritt zunächst als „trockener“ Husten in Erscheinung, mit den eintretenden Sekundärveränderungen (vermehrte Schleimsekretion durch die Drüsenhyperplasie) kann er in einen „feuchten“ Husten übergehen. Darüber hinaus kommt es zu einer verminderten Leistungsfähigkeit des Tieres.
Klinisch treten infolge ein verstärktes „tracheales Atemgeräusch“ (Stridor trachealis, zumeist in Form von Brummtönen), eine erhöhte Atemfrequenz (Tachypnoe) und zunehmende Atembeschwerden (Dyspnoe) auf. Diese treten bei Lokalisation im Halsabschnitt der Luftröhre vor allem bei der Einatmung (Inspiration), bei Manifestation im Brustteil eher bei der Ausatmung (Exspiration) auf. Die Ursache hierfür ist darin zu sehen, dass bei einer Instabilität im Halsbereich der Kollaps durch den beim Einatmen entstehenden Unterdruck verursacht wird, während eine Instabilität im Brustbereich meist ein Zusammenfallen der Luftröhre durch den beim Ausatmen auftretenden Überdruck in der Brusthöhle (bezogen auf den Druck innerhalb der Luftröhre) zur Ursache hat. Mit zunehmender Dyspnoe kommt es zu einem Sauerstoffmangel, der sich in einer Blauverfärbung (Zyanose) der Schleimhäute äußert.
Diagnose
Bei Lokalisation im Halsteil der Luftröhre lässt sich die Erweichung der Knorpelspangen bereits durch Abtasten (Palpation) feststellen, wobei meist auch Husten ausgelöst wird. Weitere Hinweise kann die Röntgendarstellung der Luftröhre liefern. Die eindeutige Diagnose ist jedoch nur durch die Endoskopie zu erbringen.
Differentialdiagnosen
Abzugrenzen sind vor allem entzündliche bedingte Erkrankungen der Luftröhre (Tracheitis) durch Infektionen oder Allergien. Auch Fremdkörper in und Tumoren der Luftröhre können Verengungen und tracheale Atemgeräusche hervorrufen. Schließlich kann eine Verengung der Luftröhre auch durch Kompression von außen durch Vergrößerung benachbarter Organe zustande kommen. Hier kommen insbesondere der Luftröhre benachbarte Organe wie die tiefen Hals- und die vorderen und mittleren mediastinalen Lymphknoten (Lnn. cervicales profundi und mediastinales craniales et medii), die Schilddrüse und die Epithelkörperchen in Frage. Auch Abszesse und Blutergüsse in der Nachbarschaft der Luftröhre können zu einer Kompression von außen führen.
Relativ häufig tritt eine Kompression der Endaufzweigung der Luftröhre infolge einer Vergrößerung des linken Vorhofes des Herzens auf, dessen Ursache wiederum eine Insuffizienz der Mitralklappe des Herzens ist (Klappenendokardiose). Hierbei wird die Symptomatik neben der Verengung des Lumens maßgeblich durch die dauernde mechanische Reizung der Luftröhre infolge der Herzbewegung verstärkt.
Bei einigen Hunderassen (Englische Bulldogge, Boston Terrier) muss auch an eine erblich bedingte Wachstumsstörung der Luftröhre (Trachea-Hypoplasie) in Betracht gezogen werden.
Therapie
Eine kausale Therapie ist nicht möglich.
Konservativ kann die Krankheit bei den meisten Hunden relativ lange beherrscht werden. Einfache Maßnahmen, wie die Nutzung eines Brustgeschirrs statt eines Halsbandes, das Vermeiden übermäßiger körperlicher Anstrengungen und Vermeidung von Übergewichtigkeit können durch den Hundehalter selbst durchgeführt werden.
Zur Linderung der Symptome können hustenstillende Mittel, Bronchodilatatoren (z. B. Theophyllin) oder kurzzeitig auch entzündungshemmende Glukokortikoide eingesetzt werden. Ein weiterer konservativer Therapieansatz ist der Einsatz von Parasympatholytika (Atropin, auch in Kombination mit Diphenoxylat, z. B. Lomotil®).
Im experimentellen Stadium ist das Einsetzen von ursprünglich für die Humanmedizin entwickelten Stents, die eine passive Stützung der Luftröhre gewährleisten.
Obwohl dieses Verfahren als derzeit effektivste Behandlungsmethode der Erkrankung angesehen wird (bei knapp 70 Prozent der Patienten wird nach dem Eingriff eine deutliche klinische Verbesserung festgestellt), konnte es sich noch nicht als Standardtherapie etablieren. Hauptursache hierfür sind die hohen Kosten des Stents; außerdem können Komplikationen auftreten, wenn die nicht von der Endoprothese stabilisierten Anteile der Luftröhre und Hauptbronchien kollabieren. Bei einigen Tieren tritt infolge der Ansammlung von Sekret im Bereich nicht vollständig an der Luftröhrenwand anliegender Stentanteile auch weiterhin Husten auf. Selten kommt es zu einer Beeinträchtigung der Atmung durch überschießende Bildung von Granulationsgewebe; diese Symptome scheinen jedoch mit der Gabe von Glukokortikoiden behebbar zu sein. Einzelberichte erwähnen das Kollabieren eines Stents mit anschließender Verengung der Luftröhre.
Als chirurgische Alternative zum Stent ist die Fixierung der Luftröhre durch ein um die Luftröhre gelegtes Kunststoffgerüst die am weiteseten verbreitete Vorgehensweise. Als weitere Möglichkeiten sind Eingriffe an der obere liegenden Membran der Luftröhre sowie direkte Manipulationen der Knorpelspangen beschrieben. Diesen Techniken gemeinsam ist, dass sie hochgradig invasiv und chirurgisch sehr anspruchsvolle Verfahren sind.
Vorkommen und Ursachen
Ein Trachealkollaps kommt vor allem bei Haushunden, insbesondere bei zwergwüchsigen Hunderassen wie z.B. bei Yorkshireterrier, Chihuahua, Malteser, Zwergspitz und Zwergschnauzer vor. Vor allem Tiere mittleren Alters sind betroffen.
Die Ursache der Erkrankung ist bislang nicht geklärt. Vermutlich ist der Trachealkollaps genetisch bedingt (Erbkrankheit). Aber auch die Genese durch das Zusammentreffen verschiedener Faktoren (multifaktoriell) ist nicht ausgeschlossen. Die Erkrankung kann durch eine Reihe anderer Faktoren, darunter Infektionen der Atemwege, Allergien, Verengungen der Luftröhre, toxische Stäube und Dämpfe sowie Herzinsuffizienz, begünstigt werden.
Pathogenese
Die Knorpelerweichung kommt durch Veränderungen der Knorpelgrundsubstanz, vor allem durch einen verminderten Gehalt an Glykosaminoglykanen (insbesondere Chondroitinsulfat) und Glykoproteinen zustande, wodurch die Wasserbindungskapazität des Knorpels und damit dessen Elastizität sinkt. Die Zellzahl im Knorpel ist erniedrigt und die Grundsubstanz ist porös. Die Abnahme des Luftröhreninnendurchmessers geht mit einem erhöhten Strömungswiderstand der Luft einher und führt zu Turbulenzen.
Sekundär kommt es durch den Trachealkollaps zu einer Degeneration des Flimmerepithels, zu einer Hypertrophie der Drüsen in der Schleimhaut der Luftröhre, gelegentlich auch zur Bildung von Polypen.
Klinik
Das klinische Erscheinungsbild ist sehr variabel, ein Trachealkollaps kann lange symptomlos bleiben und der Grad der Verengung muss nicht mit dem Ausmaß klinischer Erscheinungen korrelieren. Die Symptome entwickeln sich allmählich und die Erkrankung schreitet langsam voran.
Eine typische Früherscheinung ist ein anfallsartiger Husten, der vor allem bei Aufregung oder stärkerer körperlicher Belastung auftritt. Auch ein Halsband kann bei starkem Zug an der Leine Druck auf die Luftröhre ausüben und somit die Hustenanfälle auslösen. Der Husten tritt zunächst als „trockener“ Husten in Erscheinung, mit den eintretenden Sekundärveränderungen (vermehrte Schleimsekretion durch die Drüsenhyperplasie) kann er in einen „feuchten“ Husten übergehen. Darüber hinaus kommt es zu einer verminderten Leistungsfähigkeit des Tieres.
Klinisch treten infolge ein verstärktes „tracheales Atemgeräusch“ (Stridor trachealis, zumeist in Form von Brummtönen), eine erhöhte Atemfrequenz (Tachypnoe) und zunehmende Atembeschwerden (Dyspnoe) auf. Diese treten bei Lokalisation im Halsabschnitt der Luftröhre vor allem bei der Einatmung (Inspiration), bei Manifestation im Brustteil eher bei der Ausatmung (Exspiration) auf. Die Ursache hierfür ist darin zu sehen, dass bei einer Instabilität im Halsbereich der Kollaps durch den beim Einatmen entstehenden Unterdruck verursacht wird, während eine Instabilität im Brustbereich meist ein Zusammenfallen der Luftröhre durch den beim Ausatmen auftretenden Überdruck in der Brusthöhle (bezogen auf den Druck innerhalb der Luftröhre) zur Ursache hat. Mit zunehmender Dyspnoe kommt es zu einem Sauerstoffmangel, der sich in einer Blauverfärbung (Zyanose) der Schleimhäute äußert.
Diagnose
Bei Lokalisation im Halsteil der Luftröhre lässt sich die Erweichung der Knorpelspangen bereits durch Abtasten (Palpation) feststellen, wobei meist auch Husten ausgelöst wird. Weitere Hinweise kann die Röntgendarstellung der Luftröhre liefern. Die eindeutige Diagnose ist jedoch nur durch die Endoskopie zu erbringen.
Differentialdiagnosen
Abzugrenzen sind vor allem entzündliche bedingte Erkrankungen der Luftröhre (Tracheitis) durch Infektionen oder Allergien. Auch Fremdkörper in und Tumoren der Luftröhre können Verengungen und tracheale Atemgeräusche hervorrufen. Schließlich kann eine Verengung der Luftröhre auch durch Kompression von außen durch Vergrößerung benachbarter Organe zustande kommen. Hier kommen insbesondere der Luftröhre benachbarte Organe wie die tiefen Hals- und die vorderen und mittleren mediastinalen Lymphknoten (Lnn. cervicales profundi und mediastinales craniales et medii), die Schilddrüse und die Epithelkörperchen in Frage. Auch Abszesse und Blutergüsse in der Nachbarschaft der Luftröhre können zu einer Kompression von außen führen.
Relativ häufig tritt eine Kompression der Endaufzweigung der Luftröhre infolge einer Vergrößerung des linken Vorhofes des Herzens auf, dessen Ursache wiederum eine Insuffizienz der Mitralklappe des Herzens ist (Klappenendokardiose). Hierbei wird die Symptomatik neben der Verengung des Lumens maßgeblich durch die dauernde mechanische Reizung der Luftröhre infolge der Herzbewegung verstärkt.
Bei einigen Hunderassen (Englische Bulldogge, Boston Terrier) muss auch an eine erblich bedingte Wachstumsstörung der Luftröhre (Trachea-Hypoplasie) in Betracht gezogen werden.
Therapie
Eine kausale Therapie ist nicht möglich.
Konservativ kann die Krankheit bei den meisten Hunden relativ lange beherrscht werden. Einfache Maßnahmen, wie die Nutzung eines Brustgeschirrs statt eines Halsbandes, das Vermeiden übermäßiger körperlicher Anstrengungen und Vermeidung von Übergewichtigkeit können durch den Hundehalter selbst durchgeführt werden.
Zur Linderung der Symptome können hustenstillende Mittel, Bronchodilatatoren (z. B. Theophyllin) oder kurzzeitig auch entzündungshemmende Glukokortikoide eingesetzt werden. Ein weiterer konservativer Therapieansatz ist der Einsatz von Parasympatholytika (Atropin, auch in Kombination mit Diphenoxylat, z. B. Lomotil®).
Im experimentellen Stadium ist das Einsetzen von ursprünglich für die Humanmedizin entwickelten Stents, die eine passive Stützung der Luftröhre gewährleisten.
Obwohl dieses Verfahren als derzeit effektivste Behandlungsmethode der Erkrankung angesehen wird (bei knapp 70 Prozent der Patienten wird nach dem Eingriff eine deutliche klinische Verbesserung festgestellt), konnte es sich noch nicht als Standardtherapie etablieren. Hauptursache hierfür sind die hohen Kosten des Stents; außerdem können Komplikationen auftreten, wenn die nicht von der Endoprothese stabilisierten Anteile der Luftröhre und Hauptbronchien kollabieren. Bei einigen Tieren tritt infolge der Ansammlung von Sekret im Bereich nicht vollständig an der Luftröhrenwand anliegender Stentanteile auch weiterhin Husten auf. Selten kommt es zu einer Beeinträchtigung der Atmung durch überschießende Bildung von Granulationsgewebe; diese Symptome scheinen jedoch mit der Gabe von Glukokortikoiden behebbar zu sein. Einzelberichte erwähnen das Kollabieren eines Stents mit anschließender Verengung der Luftröhre.
Als chirurgische Alternative zum Stent ist die Fixierung der Luftröhre durch ein um die Luftröhre gelegtes Kunststoffgerüst die am weiteseten verbreitete Vorgehensweise. Als weitere Möglichkeiten sind Eingriffe an der obere liegenden Membran der Luftröhre sowie direkte Manipulationen der Knorpelspangen beschrieben. Diesen Techniken gemeinsam ist, dass sie hochgradig invasiv und chirurgisch sehr anspruchsvolle Verfahren sind.
eingestellt von: ise; Quellen- und Bildnachweis
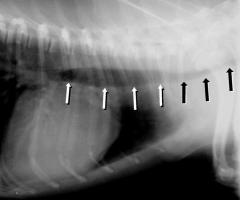
Von-Willebrand-Krankheit
Die von-Willebrand-Krankheit ist eine Blutgerinnungsstörung, die durch einen erblichen Mangel an von-Willebrand-Faktor (syn. Faktor-VIII bezogenes Antigen) ausgelöst wird. Sie ist die häufigste Störung der Blutgerinnung beim Hund.
Symptome
Zu den Symptomen gehören Zahnfleischbluten, Nasenbluten und Hämaturie. Viele der betroffenen Tiere zeigen allerdings erst nach Injektionen, Blutentnahmen oder chirurgischen Eingriffen eine verstärkte Blutungstendenz (beispielsweise bei der Entfernung der Wolfskrallen der Welpen).
Pathophysiologie
Bei gesunden Tieren befindet sich der Faktor auf der Innenwand der Blutgefäße und in den Blutplättchen. Kommt es zu einer Verletzung, bindet der Faktor an die dadurch freigelegten Kollagene und bindet die Blutplättchen an sich, wodurch die zelluläre Hämostase ausgelöst wird und die Blutplättchen mit dem Verschluss der Wunde beginnen (Thrombozytenaggregation).
Ist die Bildung des von-Willebrand-Faktors durch eine Mutation des zugehörigen Gens gestört, kann das Tier keinen oder zu wenig funktionierenden von-Willebrand-Faktor bilden. Dadurch kommt es bei einer Verletzung nicht zur Adhäsion und Aggregation der Blutplättchen, und die Blutgerinnung bleibt auf die plasmatische Komponente der Hämostase beschränkt. Dies führt je nach Schweregrad des Mangels bei Verletzungen zu einer verlängerten Blutungszeit und dadurch zu einem höheren Blutverlust, der durch Schock oder Anämie tödlich enden kann.
Klinik
Signalement
Die Krankheit ist bei mehr als fünfzig verschiedenen Hunderassen beschrieben. Besonders häufig (Prävalenz zwischen 10 und 70 %) kommt sie bei Dobermann, Deutscher Schäferhund, Golden Retriever, Zwergschnauzer, Welsh Corgi Pembroke, Shetland Sheepdog, Basset Hound, Scottish Terrier, Großpudel und Manchester Terrier vor.
Das Diagnosealter ist je nach Schwere der Erkrankung sehr variabel und kann vom Neugeboren bis hin zu alten Individuen reichen, wobei schwere Fälle normalerweise bei jüngeren Tieren diagnostiziert werden.
Diagnose
Der Verdacht auf eine von-Willebrand-Erkrankung ergibt sich bei Tieren mit klinischen Blutgerinnungsstörungen, bei denen eine Blutuntersuchung normale Blutplättchenwerte ergibt und bei denen Tests auf Gerinnungsfaktoren der plasmatischen Gerinnungskaskade normal ausfallen. Die Schleimhautblutungszeit ist fast immer verlängert. Ein negativer oder stark verminderter Bluttest auf von-Willebrand-Faktor (mittels ELISA) ist diagnostisch.
Differentialdiagnostisch kommen Thrombozytopenie oder Defekte der Blutplättchen in Betracht. Gelegentlich haben betroffene Tiere außerdem einen Mangel an Faktor VIII und dadurch eine verlängerte Partielle Thromboplastinzeit.
Therapie und Prognose
Die von-Willebrand-Krankheit ist nicht heilbar. Starke Blutungen können mit einer Blut- oder Plasmatransfusion behandelt werden. Vor Operationen ist eine präventive Gabe von Desmopressin empfehlenswert, das die Blutungstendenz verringern kann.
Neuere Studien haben ergeben, dass die Gabe von Levothyroxin nicht empfehlenswert ist und die Blutungstendenz sogar verschlimmern kann, so dass aus heutiger Sicht von dieser Therapie abgeraten werden muss.
Je nach Schweregrad der Krankheit variiert die Prognose von exzellent bis infaust.
Genetik und Zuchthygiene
Die von-Willebrand-Erkrankung kann zwei verschiedenen Erbgängen folgen:
Die häufigere Form folgt einem einfach autosomal intermediären Erbgang, bei dem die für das defekte Allel heterozygoten Tiere eine weniger stark erhöhte Blutungstendenz haben als die homozygoten. Der Schweregrad dieser Form kann zwischen einzelnen Rassen stark variieren.
Die seltenere Form vererbt sich einfach autosomal rezessiv: Hier erkranken nur die für das defekte Allel homozygoten Tiere, während die heterozygoten klinisch normale Träger sind. Für das defekte Allel homozygote Tiere sterben in der Regel vor Erreichen der Geschlechtsreife (Letalfaktor).
Im Gegensatz zu vielen anderen Krankheiten mit erhöhter Blutungsneigung (Hämophilie) wird die von-Willebrand-Krankheit nicht geschlechtsgebunden vererbt: Männliche und weibliche Tiere sind gleich häufig betroffen.
Betroffene Tiere sollten unter Berücksichtigung der klinischen Aspekte der Krankheit in der jeweiligen Rasse nicht zur Zucht verwendet werden. Gentests sind für verschiedene Rassen verfügbar, wobei die genauen Mutationen je nach Rasse variieren können und die klinischen Symptome nur bedingt mit dem Genotyp korrelieren.
Symptome
Zu den Symptomen gehören Zahnfleischbluten, Nasenbluten und Hämaturie. Viele der betroffenen Tiere zeigen allerdings erst nach Injektionen, Blutentnahmen oder chirurgischen Eingriffen eine verstärkte Blutungstendenz (beispielsweise bei der Entfernung der Wolfskrallen der Welpen).
Pathophysiologie
Bei gesunden Tieren befindet sich der Faktor auf der Innenwand der Blutgefäße und in den Blutplättchen. Kommt es zu einer Verletzung, bindet der Faktor an die dadurch freigelegten Kollagene und bindet die Blutplättchen an sich, wodurch die zelluläre Hämostase ausgelöst wird und die Blutplättchen mit dem Verschluss der Wunde beginnen (Thrombozytenaggregation).
Ist die Bildung des von-Willebrand-Faktors durch eine Mutation des zugehörigen Gens gestört, kann das Tier keinen oder zu wenig funktionierenden von-Willebrand-Faktor bilden. Dadurch kommt es bei einer Verletzung nicht zur Adhäsion und Aggregation der Blutplättchen, und die Blutgerinnung bleibt auf die plasmatische Komponente der Hämostase beschränkt. Dies führt je nach Schweregrad des Mangels bei Verletzungen zu einer verlängerten Blutungszeit und dadurch zu einem höheren Blutverlust, der durch Schock oder Anämie tödlich enden kann.
Klinik
Signalement
Die Krankheit ist bei mehr als fünfzig verschiedenen Hunderassen beschrieben. Besonders häufig (Prävalenz zwischen 10 und 70 %) kommt sie bei Dobermann, Deutscher Schäferhund, Golden Retriever, Zwergschnauzer, Welsh Corgi Pembroke, Shetland Sheepdog, Basset Hound, Scottish Terrier, Großpudel und Manchester Terrier vor.
Das Diagnosealter ist je nach Schwere der Erkrankung sehr variabel und kann vom Neugeboren bis hin zu alten Individuen reichen, wobei schwere Fälle normalerweise bei jüngeren Tieren diagnostiziert werden.
Diagnose
Der Verdacht auf eine von-Willebrand-Erkrankung ergibt sich bei Tieren mit klinischen Blutgerinnungsstörungen, bei denen eine Blutuntersuchung normale Blutplättchenwerte ergibt und bei denen Tests auf Gerinnungsfaktoren der plasmatischen Gerinnungskaskade normal ausfallen. Die Schleimhautblutungszeit ist fast immer verlängert. Ein negativer oder stark verminderter Bluttest auf von-Willebrand-Faktor (mittels ELISA) ist diagnostisch.
Differentialdiagnostisch kommen Thrombozytopenie oder Defekte der Blutplättchen in Betracht. Gelegentlich haben betroffene Tiere außerdem einen Mangel an Faktor VIII und dadurch eine verlängerte Partielle Thromboplastinzeit.
Therapie und Prognose
Die von-Willebrand-Krankheit ist nicht heilbar. Starke Blutungen können mit einer Blut- oder Plasmatransfusion behandelt werden. Vor Operationen ist eine präventive Gabe von Desmopressin empfehlenswert, das die Blutungstendenz verringern kann.
Neuere Studien haben ergeben, dass die Gabe von Levothyroxin nicht empfehlenswert ist und die Blutungstendenz sogar verschlimmern kann, so dass aus heutiger Sicht von dieser Therapie abgeraten werden muss.
Je nach Schweregrad der Krankheit variiert die Prognose von exzellent bis infaust.
Genetik und Zuchthygiene
Die von-Willebrand-Erkrankung kann zwei verschiedenen Erbgängen folgen:
Die häufigere Form folgt einem einfach autosomal intermediären Erbgang, bei dem die für das defekte Allel heterozygoten Tiere eine weniger stark erhöhte Blutungstendenz haben als die homozygoten. Der Schweregrad dieser Form kann zwischen einzelnen Rassen stark variieren.
Die seltenere Form vererbt sich einfach autosomal rezessiv: Hier erkranken nur die für das defekte Allel homozygoten Tiere, während die heterozygoten klinisch normale Träger sind. Für das defekte Allel homozygote Tiere sterben in der Regel vor Erreichen der Geschlechtsreife (Letalfaktor).
Im Gegensatz zu vielen anderen Krankheiten mit erhöhter Blutungsneigung (Hämophilie) wird die von-Willebrand-Krankheit nicht geschlechtsgebunden vererbt: Männliche und weibliche Tiere sind gleich häufig betroffen.
Betroffene Tiere sollten unter Berücksichtigung der klinischen Aspekte der Krankheit in der jeweiligen Rasse nicht zur Zucht verwendet werden. Gentests sind für verschiedene Rassen verfügbar, wobei die genauen Mutationen je nach Rasse variieren können und die klinischen Symptome nur bedingt mit dem Genotyp korrelieren.
eingestellt von: ise; Quellennachweis
Neuen Eintrag erstellen  Hundefutter-LexikonEin ständiges Thema: Was ist das beste Futter für meinen Hund!Nassfutter oder Trockenfutter, für wellches Alter geeignet und mit welchen Inhaltsstoffen, das sind Fragen mit denen sich der verantwortungsbewusste Tierhalter in der Regel
Hundefutter-LexikonEin ständiges Thema: Was ist das beste Futter für meinen Hund!Nassfutter oder Trockenfutter, für wellches Alter geeignet und mit welchen Inhaltsstoffen, das sind Fragen mit denen sich der verantwortungsbewusste Tierhalter in der Regel Einstieg in die Dooggs-Portal-Werbung | Basic-Paket!Sie suchen eine sehr günstige Möglichkeit im Dooggs-Portal zu werben?, Sie bieten Dienstleistungen zum Thema rund um den Hund an?, Dann ist unser Einsteiger-Werbe-Paket Basic
Einstieg in die Dooggs-Portal-Werbung | Basic-Paket!Sie suchen eine sehr günstige Möglichkeit im Dooggs-Portal zu werben?, Sie bieten Dienstleistungen zum Thema rund um den Hund an?, Dann ist unser Einsteiger-Werbe-Paket Basic Die mobile Trimmtante mit Herz!Das bin ich. Sarah Morich, geboren am 08.07.1991 in Herzberg am Harz. Seitdem ich mich erinnern kann, sind wir im Besitz von Hunden.2014 sagte ich mir.. warum, nicht Hobby zum Beruf gesagt, getan.. Ich machte mich selbständig als mobile
Die mobile Trimmtante mit Herz!Das bin ich. Sarah Morich, geboren am 08.07.1991 in Herzberg am Harz. Seitdem ich mich erinnern kann, sind wir im Besitz von Hunden.2014 sagte ich mir.. warum, nicht Hobby zum Beruf gesagt, getan.. Ich machte mich selbständig als mobile VMV Verband marktorientierter Verbraucher e.V.Eine günstige Haftpflichtversicherung für Ihren Hund!10 Cent pro Tag | 35.70 € im JahrMehr muss eine günstige Haftpflichtversicherung auch für Ihre Fellnase im Jahr nicht kosten, wenn Sie das überzeugende Angebot annehmen, das der
VMV Verband marktorientierter Verbraucher e.V.Eine günstige Haftpflichtversicherung für Ihren Hund!10 Cent pro Tag | 35.70 € im JahrMehr muss eine günstige Haftpflichtversicherung auch für Ihre Fellnase im Jahr nicht kosten, wenn Sie das überzeugende Angebot annehmen, das der Z.O.P.F Hundeschule in Villingen-SchwenningenTraining ohne Leckerchen?!Ja, es ist möglich, sofern der Hund sich durch andere Dinge motivieren lässt. Das können Streicheleinheiten sein, einfach nur Nähe, Spielzeug oder Dinge, die der Hund grade lieber machen möchte.Manche
Z.O.P.F Hundeschule in Villingen-SchwenningenTraining ohne Leckerchen?!Ja, es ist möglich, sofern der Hund sich durch andere Dinge motivieren lässt. Das können Streicheleinheiten sein, einfach nur Nähe, Spielzeug oder Dinge, die der Hund grade lieber machen möchte.Manche Schönhalde Tierbestattung in AlbstadtDamit Ihr Freund unvergessen bleibt!Man wünscht sich, dass man uns nie braucht...…aber wenn die Zeit kommt, dann ist es schön, jemand Erfahrenes an der Hand zu haben.Wir machen diese wichtige Arbeit mit viel Herz und Einfühlungsvermögen.Die
Schönhalde Tierbestattung in AlbstadtDamit Ihr Freund unvergessen bleibt!Man wünscht sich, dass man uns nie braucht...…aber wenn die Zeit kommt, dann ist es schön, jemand Erfahrenes an der Hand zu haben.Wir machen diese wichtige Arbeit mit viel Herz und Einfühlungsvermögen.Die „Mein Hund fürs Leben“ (ZDF)Die Redaktion der TV-Produktionsfirma „Tower Productions“ in Berlin, sucht aktuell für die zweite Staffel der Fernsehsendung „Mein Hund fürs Leben“ (ZDF) Menschen, die einen Hund aus dem Tierheim adoptieren möchten und sich auf diesem
„Mein Hund fürs Leben“ (ZDF)Die Redaktion der TV-Produktionsfirma „Tower Productions“ in Berlin, sucht aktuell für die zweite Staffel der Fernsehsendung „Mein Hund fürs Leben“ (ZDF) Menschen, die einen Hund aus dem Tierheim adoptieren möchten und sich auf diesem

DOOGGS NEWS
Hundefutter-LexikonEin ständiges Thema: Was ist das beste Futter für meinen Hund!Nassfutter oder Trockenfutter, für wellches Alter geeignet und mit welchen Inhaltsstoffen, das sind Fragen mit denen sich der verantwortungsbewusste Tierhalter in der RegelEinstieg in die Dooggs-Portal-Werbung | Basic-Paket!Sie suchen eine sehr günstige Möglichkeit im Dooggs-Portal zu werben?, Sie bieten Dienstleistungen zum Thema rund um den Hund an?, Dann ist unser Einsteiger-Werbe-Paket BasicDie mobile Trimmtante mit Herz!Das bin ich. Sarah Morich, geboren am 08.07.1991 in Herzberg am Harz. Seitdem ich mich erinnern kann, sind wir im Besitz von Hunden.2014 sagte ich mir.. warum, nicht Hobby zum Beruf gesagt, getan.. Ich machte mich selbständig als mobileVMV Verband marktorientierter Verbraucher e.V.Eine günstige Haftpflichtversicherung für Ihren Hund!10 Cent pro Tag | 35.70 € im JahrMehr muss eine günstige Haftpflichtversicherung auch für Ihre Fellnase im Jahr nicht kosten, wenn Sie das überzeugende Angebot annehmen, das derZ.O.P.F Hundeschule in Villingen-SchwenningenTraining ohne Leckerchen?!Ja, es ist möglich, sofern der Hund sich durch andere Dinge motivieren lässt. Das können Streicheleinheiten sein, einfach nur Nähe, Spielzeug oder Dinge, die der Hund grade lieber machen möchte.MancheSchönhalde Tierbestattung in AlbstadtDamit Ihr Freund unvergessen bleibt!Man wünscht sich, dass man uns nie braucht...…aber wenn die Zeit kommt, dann ist es schön, jemand Erfahrenes an der Hand zu haben.Wir machen diese wichtige Arbeit mit viel Herz und Einfühlungsvermögen.Die„Mein Hund fürs Leben“ (ZDF)Die Redaktion der TV-Produktionsfirma „Tower Productions“ in Berlin, sucht aktuell für die zweite Staffel der Fernsehsendung „Mein Hund fürs Leben“ (ZDF) Menschen, die einen Hund aus dem Tierheim adoptieren möchten und sich auf diesem


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}